 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2e7d | ||||||
|---|---|---|---|---|---|---|---|
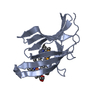
| Title | Crystal structure of a NEAT domain from Staphylococcus aureus | ||||||
 Components Components | Hypothetical protein IsdH Hypothesis Hypothesis | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / Ig-like fold | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |   Staphylococcus aureus (bacteria) Staphylococcus aureus (bacteria) | ||||||
| Method | X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.2 Å | ||||||
 Authors Authors | Suenaga, A. / Tanaka, Y. / Yao, M. / Kumagai, I. / Tanaka, I. / Tsumoto, K. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2008 Title: Structural basis for multimeric heme complexation through a specific protein-heme interaction: the case of the third neat domain of IsdH from Staphylococcus aureus Authors: Watanabe, M. / Tanaka, Y. / Suenaga, A. / Kuroda, M. / Yao, M. / Watanabe, N. / Arisaka, F. / Ohta, T. / Tanaka, I. / Tsumoto, K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2e7d.cif.gz | 61 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2e7d.ent.gz | 45.6 KB | Display | PDB format |
| PDBx/mmJSON format | 2e7d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/e7/2e7dftp://data.pdbj.org/pub/pdb/validation_reports/e7/2e7d | HTTPS FTP |
|---|
-Related structure data










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 6
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Hypothesis / heme binding protein Mass: 14854.489 Da / Num. of mol.: 2 / Fragment: NEAT domain, residue 539-664 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Staphylococcus aureus (bacteria) / Strain: Mu50 / Gene: IsdH / Plasmid: pDBHT3 / Production host: Escherichia coli (E. coli) / Strain (production host): B834(DE3) / References: UniProt: Q931P4#2: Chemical | Sulfate  Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | ChemComp-ACT / | Acetate  Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H3O2#4: Chemical | Glycerol  Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / | Water Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.99 Å3/Da / Density % sol: 58.88 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.25 Details: 0.1M sodium acetate, 2.2M ammonium sulfate, pH 4.25, VAPOR DIFFUSION, HANGING DROP, temperature 293K |
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation |
| |||||||||||||||
| Radiation wavelength |
| |||||||||||||||
| Reflection | Resolution: 2.2→50 Å / Num. obs: 18212 / % possible obs: 99.9 % / Redundancy: 11.1 % / Biso Wilson estimate: 24 Å2 / Rsym value: 0.042 | |||||||||||||||
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 10.6 % / Rsym value: 0.343 / % possible all: 100 |

- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.2→19.25 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 2476559.14 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 53.6784 Å2 / ksol: 0.352619 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 41.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→19.25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.34 Å / Rfactor Rfree error: 0.019 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
|
