 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9htv: McCP in complex with photocaged nitric oxide, 1.44 s, 0.95 microj... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9htv | ||||||
|---|---|---|---|---|---|---|---|
| Title | McCP in complex with photocaged nitric oxide, 1.44 s, 0.95 microjoule, SSX | ||||||
 Components Components | Cytochrome c | ||||||
 Keywords Keywords | METAL BINDING PROTEIN / McCP / heme / gas binding / NO | ||||||
| Function / homology | Cytochrome P460 / Cytochrome P460 superfamily / Cytochrome P460 / metal ion binding / HEME C / NITRIC OXIDE / Cytochrome c Function and homology information Function and homology information | ||||||
| Biological species |  Methylococcus capsulatus str. Bath (bacteria) Methylococcus capsulatus str. Bath (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Smyth, P. / Williams, L.J. / Hough, M.A. / Worrall, J.A.R. / Owen, R.L. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Iucrj / Year: 2025 Title: Time-resolved serial synchrotron and serial femtosecond crystallography of heme proteins using photocaged nitric oxide. Authors: Smyth, P. / Jaho, S. / Williams, L.J. / Karras, G. / Fitzpatrick, A. / Thompson, A.J. / Battah, S. / Axford, D. / Horrell, S. / Lucic, M. / Ishihara, K. / Kataoka, M. / Matsuura, H. / ...Authors: Smyth, P. / Jaho, S. / Williams, L.J. / Karras, G. / Fitzpatrick, A. / Thompson, A.J. / Battah, S. / Axford, D. / Horrell, S. / Lucic, M. / Ishihara, K. / Kataoka, M. / Matsuura, H. / Shimba, K. / Tono, K. / Tosha, T. / Sugimoto, H. / Owada, S. / Hough, M.A. / Worrall, J.A.R. / Owen, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9htv.cif.gz | 76.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9htv.ent.gz | 53.7 KB | Display | PDB format |
| PDBx/mmJSON format | 9htv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ht/9htvftp://data.pdbj.org/pub/pdb/validation_reports/ht/9htv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9hl1C  9ho7C  9hqtC  9hs8C  9htcC  9httC  9hu1C  9hxxC  9hyvC  9hyzC  9i4qC  9i4sC  9i4uC  9i6gC  9ia9C  9iaaC  9q86C  9qmeC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 15244.063 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methylococcus capsulatus str. Bath (bacteria)Gene: ccp, MCA2394 / Plasmid: pBluescript SK(+) / Production host: #2: Chemical |   Mass: 618.503 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H34FeN4O4 Mass: 618.503 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H34FeN4O4#3: Chemical | ChemComp-ZN / |   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn#4: Chemical | 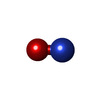  Mass: 30.006 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: NO / Feature type: SUBJECT OF INVESTIGATION Mass: 30.006 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: NO / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 68 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 68 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.97 Å3/Da / Density % sol: 63.6 % / Description: Cubes of approx 30 um grew within 24 h. |
|---|---|
| Crystal grow | Temperature: 291 K / Method: batch mode Details: Final concentrations: 20 mg/mL protein, 50 mM HEPES pH 7.5, 34 % (v/v) polyethylene glycol 550, 500 mM MES pH 6.5, 5 mM ZnSO4. |
-Data collection
| Diffraction | Mean temperature: 293 K / Serial crystal experiment: Y | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond  / Beamline: I24 / Wavelength: 1 Å / Beamline: I24 / Wavelength: 1 Å | ||||||||||||||||||
| Detector | Type: DECTRIS PILATUS3 6M / Detector: PIXEL / Date: Sep 30, 2021 | ||||||||||||||||||
| Radiation | Monochromator: Cryocooled double crystal monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||
| Reflection twin |
| ||||||||||||||||||
| Reflection | Resolution: 1.8→76.064 Å / Num. obs: 38471 / % possible obs: 100 % / Redundancy: 59.7 % / CC1/2: 0.993 / R split: 0.111 / Net I/σ(I): 6.3 | ||||||||||||||||||
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 47.1 % / Mean I/σ(I) obs: 0.7 / Num. unique obs: 1909 / CC1/2: 0.399 / R split: 0.943 / % possible all: 100 | ||||||||||||||||||
| Serial crystallography measurement | Collection time total: 0.3 hours / Collimation: Kirkpatrick-Baez mirrors / Source size: 64 µm2 | ||||||||||||||||||
| Serial crystallography sample delivery | Method: fixed target | ||||||||||||||||||
| Serial crystallography sample delivery fixed target | Description: Oxford silicon chip / Motion control: Geobrick and Smaract / Sample dehydration prevention: 6 um Mylar, 6 um EVAL | ||||||||||||||||||
| Serial crystallography data reduction | Frames total: 25600 / Lattices indexed: 7713 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.8→76.064 Å / Cor.coef. Fo:Fc: 0.976 / Cor.coef. Fo:Fc free: 0.968 / SU B: 2.763 / SU ML: 0.077 / Cross valid method: FREE R-VALUE / ESU R: 0.087 / ESU R Free: 0.084 / Details: Hydrogens have not been used
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.963 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→76.064 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20 / % reflection obs: 100 %
|
