Mass: 18.015 Da / Num. of mol.: 127 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has ligand of interest
Y
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.21 Å3/Da / Density % sol: 44 %
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop Details: ATX was incubated with each screened compound at a 1:10 (protein:compound) ratio for at least 30 minutes. Crystals were grown for at least 7 days in a 24-well optimization screen: 18 to 20% ...Details: ATX was incubated with each screened compound at a 1:10 (protein:compound) ratio for at least 30 minutes. Crystals were grown for at least 7 days in a 24-well optimization screen: 18 to 20% PEG 3350, 0.1 to 0.4 M NaSCN, and 0.1 to 0.4 M NH4I.
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Resolution: 2.9→3.08 Å / Redundancy: 1.7 % / Num. unique obs: 3033 / CC1/2: 0.603 / % possible all: 96.9
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0419
refinement
Aimless
1.12.15
datascaling
MOLREP
11.7.03
phasing
Coot
0.9.8.92
modelbuilding
MolProbity
4.5.2
modelbuilding
XDS
datareduction
PDB-REDO
refinement
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.9→44.166 Å / Cor.coef. Fo:Fc: 0.916 / Cor.coef. Fo:Fc free: 0.843 / SU B: 57.94 / SU ML: 0.491 / Cross valid method: THROUGHOUT / ESU R Free: 0.491 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2681
964
5.147 %
RANDOM
Rwork
0.2066
17766
-
-
all
0.21
-
-
-
obs
-
18730
96.846 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 52.241 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-5.382 Å2
4.93 Å2
-0.435 Å2
2-
-
-0.862 Å2
2.522 Å2
3-
-
-
2.759 Å2
Refinement step
Cycle: LAST / Resolution: 2.9→44.166 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
6287
0
137
127
6551
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.004
0.016
6784
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.016
6058
X-RAY DIFFRACTION
r_angle_refined_deg
0.865
1.784
9224
X-RAY DIFFRACTION
r_angle_other_deg
0.333
1.565
14015
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
6.7
5.291
824
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
23.151
10
52
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
12.367
10
1076
X-RAY DIFFRACTION
r_dihedral_angle_6_deg
11.731
10
313
X-RAY DIFFRACTION
r_chiral_restr
0.042
0.2
925
X-RAY DIFFRACTION
r_gen_planes_refined
0.003
0.02
7821
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
1595
X-RAY DIFFRACTION
r_nbd_refined
0.175
0.2
1503
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.174
0.2
6286
X-RAY DIFFRACTION
r_nbtor_refined
0.17
0.2
3239
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.088
0.2
3271
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.115
0.2
229
X-RAY DIFFRACTION
r_metal_ion_refined
0.126
0.2
2
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.144
0.2
12
X-RAY DIFFRACTION
r_nbd_other
0.201
0.2
57
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.146
0.2
6
X-RAY DIFFRACTION
r_mcbond_it
0.993
2.232
3118
X-RAY DIFFRACTION
r_mcbond_other
0.983
2.228
3110
X-RAY DIFFRACTION
r_mcangle_it
1.752
4.007
3881
X-RAY DIFFRACTION
r_mcangle_other
1.752
4.008
3882
X-RAY DIFFRACTION
r_scbond_it
0.943
2.336
3666
X-RAY DIFFRACTION
r_scbond_other
0.943
2.338
3667
X-RAY DIFFRACTION
r_scangle_it
1.57
4.265
5342
X-RAY DIFFRACTION
r_scangle_other
1.57
4.266
5343
X-RAY DIFFRACTION
r_lrange_it
4.741
26.788
28709
X-RAY DIFFRACTION
r_lrange_other
4.728
26.781
28688
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
2.9-2.975
0.411
64
0.311
1328
0.315
1435
0.891
0.945
97.0035
0.308
2.975-3.056
0.324
63
0.313
1289
0.313
1398
0.936
0.943
96.7096
0.31
3.056-3.144
0.337
60
0.296
1221
0.298
1326
0.928
0.953
96.6063
0.288
3.144-3.241
0.332
75
0.272
1194
0.276
1317
0.935
0.961
96.3554
0.263
3.241-3.346
0.326
75
0.24
1197
0.245
1319
0.95
0.97
96.4367
0.233
3.346-3.463
0.281
58
0.234
1087
0.237
1191
0.95
0.97
96.1377
0.223
3.463-3.593
0.321
55
0.23
1076
0.235
1186
0.942
0.973
95.3626
0.215
3.593-3.739
0.28
67
0.219
1028
0.223
1148
0.958
0.975
95.3833
0.206
3.739-3.903
0.249
60
0.195
1000
0.198
1091
0.966
0.981
97.1586
0.185
3.903-4.092
0.237
51
0.178
987
0.181
1059
0.964
0.983
98.017
0.171
4.092-4.312
0.17
48
0.157
932
0.157
1000
0.981
0.987
98
0.151
4.312-4.571
0.219
40
0.147
860
0.15
928
0.967
0.988
96.9828
0.146
4.571-4.882
0.247
40
0.145
835
0.15
894
0.974
0.988
97.8747
0.145
4.882-5.268
0.241
36
0.153
764
0.157
821
0.963
0.989
97.4421
0.15
5.268-5.763
0.233
46
0.196
693
0.198
758
0.971
0.981
97.4934
0.192
5.763-6.429
0.297
41
0.202
641
0.208
697
0.946
0.977
97.8479
0.205
6.429-7.398
0.29
24
0.197
551
0.201
598
0.95
0.978
96.1538
0.199
7.398-8.998
0.206
25
0.16
503
0.162
535
0.977
0.985
98.6916
0.166
8.998-12.469
0.163
24
0.151
367
0.152
399
0.981
0.987
97.995
0.159
12.469-44.166
0.313
12
0.235
212
0.238
235
0.957
0.96
95.3192
0.261
Refinement TLS params.
Method: refined / Origin x: 0.5439 Å / Origin y: 0.4988 Å / Origin z: -0.2817 Å
11
12
13
21
22
23
31
32
33
T
0.5619 Å2
-0.3575 Å2
0.1457 Å2
-
0.2371 Å2
-0.0925 Å2
-
-
0.0442 Å2
L
0.8844 °2
0.3494 °2
-0.6077 °2
-
0.4747 °2
-0.3843 °2
-
-
1.1911 °2
S
0.0398 Å °
0.0562 Å °
0.0282 Å °
0.0147 Å °
0.0338 Å °
0.0414 Å °
0.0003 Å °
-0.0248 Å °
-0.0736 Å °
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection
Auth asym-ID
Auth seq-ID
1
X-RAY DIFFRACTION
1
ALL
A
56 - 859
2
X-RAY DIFFRACTION
1
B
1 - 5
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Netherlands, 1items
Netherlands, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj

 Assembly
Assembly
 Homo sapiens (human)
Homo sapiens (human)



 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 126.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: I
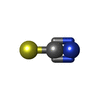
Mass: 126.904 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: I Mass: 58.082 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: CNS
Mass: 58.082 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: CNS Sample preparation
Sample preparation / Beamline: MASSIF-1 / Wavelength: 0.965459 Å
/ Beamline: MASSIF-1 / Wavelength: 0.965459 Å Processing
Processing