Mass: 18.015 Da / Num. of mol.: 368 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has ligand of interest
Y
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.29 Å3/Da / Density % sol: 46.4 %
Crystal grow
Temperature: 293 K / Method: vapor diffusion, hanging drop Details: ATX was incubated with each screened compound at a 1:10 (protein:compound) ratio for at least 30 minutes. Crystals were grown for at least 7 days in a 24-well optimization screen: 18 to 20% ...Details: ATX was incubated with each screened compound at a 1:10 (protein:compound) ratio for at least 30 minutes. Crystals were grown for at least 7 days in a 24-well optimization screen: 18 to 20% PEG 3350, 0.1 to 0.4 M NaSCN, and 0.1 to 0.4 M NH4I.
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Resolution: 2→2.05 Å / Redundancy: 1.7 % / Num. unique obs: 3821 / CC1/2: 0.588 / % possible all: 94.4
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0419
refinement
Aimless
1.12.15
datascaling
MOLREP
11.7.03
phasing
Coot
0.9.8.92
modelbuilding
MolProbity
4.5.2
modelbuilding
XDS
datareduction
PDB-REDO
refinement
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→43.19 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.94 / SU B: 11.11 / SU ML: 0.14 / Cross valid method: THROUGHOUT / ESU R: 0.199 / ESU R Free: 0.168 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2175
2571
4.935 %
RANDOM
Rwork
0.1709
49531
-
-
all
0.173
-
-
-
obs
-
52102
95.998 %
-
Solvent computation
Ion probe radii: 0.7 Å / Shrinkage radii: 0.7 Å / VDW probe radii: 1.1 Å / Solvent model: MASK BULK SOLVENT
Displacement parameters
Biso mean: 31.226 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-0.263 Å2
0.065 Å2
0.689 Å2
2-
-
-0.355 Å2
-0.166 Å2
3-
-
-
0.434 Å2
Refinement step
Cycle: LAST / Resolution: 2→43.19 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
6339
0
229
368
6936
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.013
0.016
6727
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.016
6009
X-RAY DIFFRACTION
r_angle_refined_deg
1.382
1.783
9066
X-RAY DIFFRACTION
r_angle_other_deg
0.484
1.567
13921
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
9.823
5.283
831
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
14.969
10
11
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
14.076
10
1089
X-RAY DIFFRACTION
r_dihedral_angle_6_deg
16.526
10
314
X-RAY DIFFRACTION
r_chiral_restr
0.072
0.2
930
X-RAY DIFFRACTION
r_gen_planes_refined
0.006
0.02
7696
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
1557
X-RAY DIFFRACTION
r_nbd_refined
0.213
0.2
1432
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.193
0.2
5828
X-RAY DIFFRACTION
r_nbtor_refined
0.185
0.2
3231
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.083
0.2
3412
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.166
0.2
340
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_other
0.028
0.2
2
X-RAY DIFFRACTION
r_metal_ion_refined
0.14
0.2
8
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.165
0.2
16
X-RAY DIFFRACTION
r_nbd_other
0.232
0.2
59
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.082
0.2
4
X-RAY DIFFRACTION
r_mcbond_it
5.636
1.804
3182
X-RAY DIFFRACTION
r_mcbond_other
5.096
1.78
3142
X-RAY DIFFRACTION
r_mcangle_it
6.298
3.181
3924
X-RAY DIFFRACTION
r_mcangle_other
6.297
3.182
3925
X-RAY DIFFRACTION
r_scbond_it
8.567
2.387
3545
X-RAY DIFFRACTION
r_scbond_other
8.568
2.387
3545
X-RAY DIFFRACTION
r_scangle_it
11.042
4.055
5142
X-RAY DIFFRACTION
r_scangle_other
11.031
4.056
5142
X-RAY DIFFRACTION
r_lrange_it
12.424
23.103
30048
X-RAY DIFFRACTION
r_lrange_other
12.431
23.03
29942
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
2-2.052
0.309
179
0.274
3634
X-RAY DIFFRACTION
94.4279
2.052-2.108
0.303
190
0.262
3459
X-RAY DIFFRACTION
93.6842
2.108-2.169
0.273
185
0.231
3465
X-RAY DIFFRACTION
95.375
2.169-2.235
0.273
170
0.222
3405
X-RAY DIFFRACTION
96.1797
2.235-2.309
0.247
180
0.197
3236
X-RAY DIFFRACTION
96.5517
2.309-2.389
0.235
181
0.188
3107
X-RAY DIFFRACTION
96.028
2.389-2.479
0.24
160
0.173
3074
X-RAY DIFFRACTION
96.25
2.479-2.58
0.234
154
0.165
2954
X-RAY DIFFRACTION
96.4618
2.58-2.694
0.263
135
0.163
2806
X-RAY DIFFRACTION
95.8605
2.694-2.825
0.224
128
0.147
2723
X-RAY DIFFRACTION
96.2525
2.825-2.978
0.196
127
0.141
2532
X-RAY DIFFRACTION
94.8626
2.978-3.157
0.176
133
0.13
2408
X-RAY DIFFRACTION
95.8145
3.157-3.374
0.175
126
0.132
2286
X-RAY DIFFRACTION
97.731
3.374-3.642
0.196
110
0.138
2137
X-RAY DIFFRACTION
96.9789
3.642-3.987
0.182
76
0.141
1999
X-RAY DIFFRACTION
97.3721
3.987-4.453
0.138
98
0.133
1784
X-RAY DIFFRACTION
96.8605
4.453-5.133
0.191
93
0.144
1558
X-RAY DIFFRACTION
97.2893
5.133-6.265
0.247
68
0.208
1324
X-RAY DIFFRACTION
96.1326
6.265-8.768
0.282
61
0.222
1043
X-RAY DIFFRACTION
98.0462
8.768-43.19
0.269
17
0.266
597
X-RAY DIFFRACTION
96.8454
Refinement TLS params.
Method: refined / Origin x: 0.6063 Å / Origin y: -0.0992 Å / Origin z: 1.0771 Å
11
12
13
21
22
23
31
32
33
T
0.0643 Å2
-0.0623 Å2
-0.0312 Å2
-
0.0609 Å2
0.0297 Å2
-
-
0.0155 Å2
L
0.4458 °2
0.0856 °2
0.1424 °2
-
0.6284 °2
0.1155 °2
-
-
0.7726 °2
S
0.0066 Å °
-0.0022 Å °
-0.0057 Å °
0.0088 Å °
0.0085 Å °
-0.0176 Å °
0.0061 Å °
0.0065 Å °
-0.0151 Å °
Refinement TLS group
Selection: ALL
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Netherlands, 1items
Netherlands, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj

 Assembly
Assembly
 Homo sapiens (human)
Homo sapiens (human)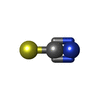






 Mass: 58.082 Da / Num. of mol.: 40 / Source method: obtained synthetically / Formula: CNS
Mass: 58.082 Da / Num. of mol.: 40 / Source method: obtained synthetically / Formula: CNS Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 126.904 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: I
Mass: 126.904 Da / Num. of mol.: 7 / Source method: obtained synthetically / Formula: I Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: PO4 Sample preparation
Sample preparation / Beamline: MASSIF-1 / Wavelength: 0.965459 Å
/ Beamline: MASSIF-1 / Wavelength: 0.965459 Å Processing
Processing