 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9dl3: Structure of proline utilization A complexed with quinoline-2-car... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9dl3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of proline utilization A complexed with quinoline-2-carboxylic acid | ||||||
 Components Components | Bifunctional protein PutA | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOENZYME / ROSSMANN FOLD / PROLINE DEHYDROGENASE / ALDEHYDE DEHYDROGENASE / PROLINE CATABOLISM / SUBSTRATE CHANNELING / BIFUNCTIONAL ENZYME | ||||||
| Function / homology |  Function and homology information Function and homology informationproline dehydrogenase / proline dehydrogenase activity / L-glutamate gamma-semialdehyde dehydrogenase / L-glutamate gamma-semialdehyde dehydrogenase activity / : / cytoplasmic side of plasma membrane / DNA-binding transcription factor activity / nucleotide binding / DNA binding Similarity search - Function | ||||||
| Biological species |  Sinorhizobium meliloti (bacteria) Sinorhizobium meliloti (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.77 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.77 Å | ||||||
 Authors Authors | Tanner, J.J. / Meeks, K.R. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Molecules / Year: 2024 Title: Crystallographic Fragment Screening of a Bifunctional Proline Catabolic Enzyme Reveals New Inhibitor Templates for Proline Dehydrogenase and L-Glutamate-gamma-semialdehyde Dehydrogenase. Authors: Meeks, K.R. / Bogner, A.N. / Nix, J.C. / Tanner, J.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9dl3.cif.gz | 939.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9dl3.ent.gz | 759 KB | Display | PDB format |
| PDBx/mmJSON format | 9dl3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dl/9dl3ftp://data.pdbj.org/pub/pdb/validation_reports/dl/9dl3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  9dl2C  9dl4C  9dl5C  9dl6C  9dl7C  9dl8C  9dl9C  9e0aC  9e0bC  9e0cC  9e0dC  9e0eC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 131961.656 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Sinorhizobium meliloti (bacteria) / Strain: SM11 / Gene: putA, SM11_chr0102 / Production host: References: UniProt: F7X6I3, proline dehydrogenase, L-glutamate gamma-semialdehyde dehydrogenase |
|---|
-Non-polymers , 9 types, 2254 molecules 

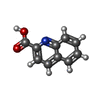






| #2: Chemical |  Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: FAD*YM#3: Chemical | ChemComp-PGE /  Mass: 150.173 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C6H14O4#4: Chemical | ChemComp-QNC /  Mass: 173.168 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H7NO2 / Feature type: SUBJECT OF INVESTIGATION Mass: 173.168 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H7NO2 / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | ChemComp-PEG /  Mass: 106.120 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 106.120 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C4H10O3#6: Chemical | ChemComp-FMT /  Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2#7: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 11 / Source method: obtained synthetically / Formula: SO4#8: Chemical | ChemComp-MG / |  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#9: Chemical | ChemComp-PG4 / |  Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM Mass: 194.226 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C8H18O5 / Comment: precipitant*YM#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 2214 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.39 Å3/Da / Density % sol: 48.56 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: Reservoir contained 0.1 M HEPES, pH 7.5, 0.1 M sodium formate, 0.1 M magnesium chloride, 0.15 M ammonium sulfate, and 22% (w/v) PEG 3350. Crystal was soaked in cryobuffer containing 50 mM ...Details: Reservoir contained 0.1 M HEPES, pH 7.5, 0.1 M sodium formate, 0.1 M magnesium chloride, 0.15 M ammonium sulfate, and 22% (w/v) PEG 3350. Crystal was soaked in cryobuffer containing 50 mM quinoline-2-carboxylic acid and 20% PEG 200 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 4.2.2 / Wavelength: 1.072156 Å |
| Detector | Type: RDI CMOS_8M / Detector: CMOS / Date: Jun 30, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.072156 Å / Relative weight: 1 |
| Reflection | Resolution: 1.77→48.53 Å / Num. obs: 412629 / % possible obs: 95.5 % / Redundancy: 3.3 % / CC1/2: 0.996 / Rmerge(I) obs: 0.079 / Rpim(I) all: 0.051 / Rrim(I) all: 0.094 / Χ2: 1 / Net I/σ(I): 12.3 |
| Reflection shell | Resolution: 1.77→1.8 Å / % possible obs: 73.2 % / Redundancy: 2.3 % / Rmerge(I) obs: 0.917 / Num. measured all: 20177 / Num. unique obs: 8678 / CC1/2: 0.426 / Rpim(I) all: 0.749 / Rrim(I) all: 1.192 / Χ2: 0.55 / Net I/σ(I) obs: 0.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS / Resolution: 1.77→48.53 Å / SU ML: 0.23 / Cross valid method: FREE R-VALUE / σ(F): 0 / Phase error: 23.86 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.1 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.77→48.53 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
