 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-9api: THE S VARIANT OF HUMAN ALPHA1-ANTITRYPSIN, STRUCTURE AND IMPLICAT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 9api | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | THE S VARIANT OF HUMAN ALPHA1-ANTITRYPSIN, STRUCTURE AND IMPLICATIONS FOR FUNCTION AND METABOLISM | |||||||||
 Components Components | (ALPHA 1-ANTITRYPSIN) x 2 | |||||||||
 Keywords Keywords | PROTEINASE INHIBITOR | |||||||||
| Function / homology |  Function and homology information Function and homology informationCargo concentration in the ER / COPII-coated ER to Golgi transport vesicle / COPII-mediated vesicle transport / endoplasmic reticulum-Golgi intermediate compartment membrane / platelet alpha granule lumen / acute-phase response / Post-translational protein phosphorylation / serine-type endopeptidase inhibitor activity / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / blood coagulation ...Cargo concentration in the ER / COPII-coated ER to Golgi transport vesicle / COPII-mediated vesicle transport / endoplasmic reticulum-Golgi intermediate compartment membrane / platelet alpha granule lumen / acute-phase response / Post-translational protein phosphorylation / serine-type endopeptidase inhibitor activity / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / blood coagulation / Platelet degranulation / extracellular matrix / protease binding / ficolin-1-rich granule lumen / endoplasmic reticulum lumen / Neutrophil degranulation / Golgi apparatus / endoplasmic reticulum / : / extracellular exosome / extracellular region / identical protein binding Similarity search - Function | |||||||||
| Method |  X-RAY DIFFRACTION / Resolution: 3 Å X-RAY DIFFRACTION / Resolution: 3 Å | |||||||||
 Authors Authors | Loebermann, H. / Tokuoka, R. / Deisenhofer, J. / Huber, R. | |||||||||
 Citation Citation | Journal: Protein Eng. / Year: 1989 Title: The S variant of human alpha 1-antitrypsin, structure and implications for function and metabolism. Authors: Engh, R. / Lobermann, H. / Schneider, M. / Wiegand, G. / Huber, R. / Laurell, C.B. #1: Journal: J.Mol.Biol. / Year: 1984Title: Human Alpha1-Proteinase Inhibitor. Crystal Structure Analysis of Two Crystal Modifications, Molecular Model and Preliminary Analysis of the Implications for Function Authors: Loebermann, H. / Tokuoka, R. / Deisenhofer, J. / Huber, R. #2: Journal: Hoppe-Seyler's Z.Physiol.Chem. / Year: 1982Title: Interaction of Human Alpha1-Proteinase Inhibitor with Chymotrypsinogena and Crystallization of a Proteolytically Modified Alpha1-Proteinase Inhibitor Authors: Loebermann, H. / Lottspeich, F. / Bode, W. / Huber, R. #3: Journal: J.Biol.Chem. / Year: 1982Title: The Biosynthesis of Rat Alpha1-Antitrypsin Authors: Carlson, J. / Stenflo, J. #4: Journal: FEBS Lett. / Year: 1981Title: Human Alpha1-Antitrypsin. Carbohydrate Attachment and Sequence Homology Authors: Carrell, R.W. / Jeppsson, J.-O. / Vaughan, L. / Brennan, S.O. / Owen, M.C. / Boswell, D.R. #5: Journal: J.Biol.Chem. / Year: 1980Title: Studies on the Oligosaccharide Chains of Human Alpha1-Protease Inhibitor. II. Structure of Oligosaccharides Authors: Mega, T. / Lujan, E. / Yoshida, A. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 9api.cif.gz | 92.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb9api.ent.gz | 65.8 KB | Display | PDB format |
| PDBx/mmJSON format | 9api.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ap/9apiftp://data.pdbj.org/pub/pdb/validation_reports/ap/9api | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Atom site foot note | 1: RESIDUE PRO B 361 IS A CIS PROLINE. 2: AN OCCUPANCY AND B VALUE OF 0.0 INDICATES THAT NO SIGNIFICANT ELECTRON DENSITY WAS FOUND IN THE FINAL FOURIER MAP. |
-Components
| #1: Protein | Mass: 39114.320 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source References: UniProt: P01009 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 4139.938 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source References: UniProt: P01009 | ||||||||
| #3: Polysaccharide | 2-acetamido-2-deoxy-beta-D-glucopyranose-(1-2)-alpha-D-mannopyranose-(1-6)-[alpha-D-mannopyranose- ...2-acetamido-2-deoxy-beta-D-glucopyranose-(1-2)-alpha-D-mannopyranose-(1-6)-[alpha-D-mannopyranose-(1-3)]alpha-D-mannopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-beta-D-glucopyranose Source method: isolated from a genetically manipulated source | ||||||||
| #4: Polysaccharide | Source method: isolated from a genetically manipulated source #5: Chemical | ChemComp-CYS / | 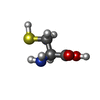  Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2S Type: L-peptide linking / Mass: 121.158 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H7NO2SCompound details | IN THIS ENTRY CHAIN IDENTIFIER *C* IS USED FOR A CYSTEINE RESIDUE WHICH FORMS A DISULFIDE BOND WITH ...IN THIS ENTRY CHAIN IDENTIFIER | Has protein modification | Y | Nonpolymer details | THE CARBOHYDRATE CHAINS PRESENTED AT THE END OF THIS ENTRY ARE NOT COMPLETE. SOME OF THE SUGAR ...THE CARBOHYDRA | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.29 Å3/Da / Density % sol: 71.32 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | *PLUS Temperature: 4 ℃ / pH: 8 / Method: vapor diffusionDetails: Loebermann, H., (1982) Hoppe-Seyler'S Z.Physiol. Chem., 363, 1377. | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Reflection | *PLUS Highest resolution: 3 Å / Lowest resolution: 9999 Å / Num. obs: 8289 / % possible obs: 47 % / Num. measured all: 12963 / Rmerge(I) obs: 0.066 |
|---|---|
| Reflection shell | *PLUS |
- Processing
Processing
| Software | Name: EREF / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Rfactor Rwork: 0.209 / Highest resolution: 3 Å Details: AN OCCUPANCY AND B VALUE OF 0.0 INDICATES THAT NO SIGNIFICANT ELECTRON DENSITY WAS FOUND IN THE FINAL FOURIER MAP. ATOMS WITH THERMAL FACTORS WHICH CALCULATE LESS THAN 6.00 ARE ASSIGNED THIS ...Details: AN OCCUPANCY AND B VALUE OF 0.0 INDICATES THAT NO SIGNIFICANT ELECTRON DENSITY WAS FOUND IN THE FINAL FOURIER MAP. ATOMS WITH THERMAL FACTORS WHICH CALCULATE LESS THAN 6.00 ARE ASSIGNED THIS VALUE. THIS IS THE LOWEST VALUE ALLOWED BY THE REFINEMENT PROGRAM. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Highest resolution: 3 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3 Å / Lowest resolution: 9999 Å / Num. reflection obs: 8289 / Rfactor obs: 0.209 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: o_angle_d |
