 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8vae: Cryogenic electron microscopy structure of human serum albumin in... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8vae | ||||||
|---|---|---|---|---|---|---|---|
| Title | Cryogenic electron microscopy structure of human serum albumin in complex with salicylic acid | ||||||
 Components Components | Serum albumin | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Human Serum Albumin / Salicylic acid | ||||||
| Function / homology |  Function and homology information Function and homology informationbilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation ...bilirubin transport / Ciprofloxacin ADME / exogenous protein binding / cellular response to calcium ion starvation / enterobactin binding / Heme biosynthesis / HDL remodeling / negative regulation of mitochondrial depolarization / Prednisone ADME / Heme degradation / molecular carrier activity / Aspirin ADME / antioxidant activity / Scavenging of heme from plasma / toxic substance binding / Recycling of bile acids and salts / platelet alpha granule lumen / fatty acid binding / cellular response to starvation / Post-translational protein phosphorylation / Cytoprotection by HMOX1 / response to nutrient levels / Regulation of Insulin-like Growth Factor (IGF) transport and uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) / Platelet degranulation / pyridoxal phosphate binding / protein-folding chaperone binding / blood microparticle / endoplasmic reticulum lumen / copper ion binding / Golgi apparatus / endoplasmic reticulum / protein-containing complex / DNA binding / : / extracellular exosome / extracellular region / identical protein binding / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.7 Å | ||||||
 Authors Authors | Catalano, C. / Lucier, K.W. / To, D. / Senko, S. / Tran, N.L. / Farwell, A.C. / Silva, S.M. / Dip, P.V. / Poweleit, N. / Scapin, G. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: J Struct Biol / Year: 2024 Title: The CryoEM structure of human serum albumin in complex with ligands. Authors: Claudio Catalano / Kyle W Lucier / Dennis To / Skerdi Senko / Nhi L Tran / Ashlyn C Farwell / Sabrina M Silva / Phat V Dip / Nicole Poweleit / Giovanna Scapin /  Abstract: Human serum albumin (HSA) is the most prevalent plasma protein in the human body, accounting for 60 % of the total plasma protein. HSA plays a major pharmacokinetic function, serving as a ...Human serum albumin (HSA) is the most prevalent plasma protein in the human body, accounting for 60 % of the total plasma protein. HSA plays a major pharmacokinetic function, serving as a facilitator in the distribution of endobiotics and xenobiotics within the organism. In this paper we report the cryoEM structures of HSA in the apo form and in complex with two ligands (salicylic acid and teniposide) at a resolution of 3.5, 3.7 and 3.4 Å, respectively. We expand upon previously published work and further demonstrate that sub-4 Å maps of ∼60 kDa proteins can be routinely obtained using a 200 kV microscope, employing standard workflows. Most importantly, these maps allowed for the identification of small molecule ligands, emphasizing the practical applicability of this methodology and providing a starting point for subsequent computational modeling and in silico optimization. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8vae.cif.gz | 113.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8vae.ent.gz | 86.2 KB | Display | PDB format |
| PDBx/mmJSON format | 8vae.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/va/8vaeftp://data.pdbj.org/pub/pdb/validation_reports/va/8vae | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  43089MC  8vacC  8vafC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |


-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 66571.219 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human)Gene: ALB, GIG20, GIG42, PRO0903, PRO1708, PRO2044, PRO2619, PRO2675, UNQ696/PRO1341 Production host: Homo sapiens (human) / References: UniProt: P02768 | ||||
|---|---|---|---|---|---|
| #2: Chemical | 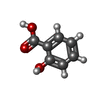  Mass: 138.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H6O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 138.121 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C7H6O3 / Feature type: SUBJECT OF INVESTIGATIONHas ligand of interest | Y | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: human serum albumin in complex with salicyl acid / Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: Homo sapiens (human) |
| Source (recombinant) | Organism: Homo sapiens (human) |
| Buffer solution | pH: 7.4 |
| Buffer component | Name: PBS |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Specimen support | Grid material: GOLD / Grid type: UltrAuFoil R1.2/1.3 |
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 95 % / Chamber temperature: 277.15 K |
- Electron microscopy imaging
Electron microscopy imaging
| Microscopy | Model: TFS GLACIOS |
|---|---|
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: SPOT SCAN FIELD EMISSION GUN / Accelerating voltage: 200 kV / Illumination mode: SPOT SCAN |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 240000 X / Nominal defocus max: 1500 nm / Nominal defocus min: 500 nm / Cs: 2.7 mm / C2 aperture diameter: 50 µm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 3.7 sec. / Electron dose: 36.43 e/Å2 / Film or detector model: FEI FALCON IV (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 9616 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||||||
| Particle selection | Num. of particles selected: 530501 | ||||||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.7 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 27064 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||||||
| Atomic model building | Protocol: RIGID BODY FIT / Space: REAL | ||||||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 1AO6 Pdb chain-ID: A / Accession code: 1AO6 / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|

