- PDB-8q61: Co-crystal structure of human AKT2 with compound 3 -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 8q61
Title
Co-crystal structure of human AKT2 with compound 3
Components
RAC-beta serine/threonine-protein kinase
Keywords
TRANSFERASE / Protein Kinase Inhibitor Complex
Function / homology
Function and homology information
retinal rod cell apoptotic process / PDE3B signalling / cellular response to high light intensity / Inhibition of TSC complex formation by PKB / positive regulation of cap-dependent translational initiation / AKT-mediated inactivation of FOXO1A / negative regulation of long-chain fatty acid import across plasma membrane / Negative regulation of the PI3K/AKT network / Activation of AKT2 / AKT phosphorylates targets in the nucleus ...retinal rod cell apoptotic process / PDE3B signalling / cellular response to high light intensity / Inhibition of TSC complex formation by PKB / positive regulation of cap-dependent translational initiation / AKT-mediated inactivation of FOXO1A / negative regulation of long-chain fatty acid import across plasma membrane / Negative regulation of the PI3K/AKT network / Activation of AKT2 / AKT phosphorylates targets in the nucleus / positive regulation of glucose metabolic process / positive regulation of fatty acid beta-oxidation / RUNX2 regulates genes involved in cell migration / mammary gland epithelial cell differentiation / RAB GEFs exchange GTP for GDP on RABs / glycogen biosynthetic process / peripheral nervous system myelin maintenance / AKT phosphorylates targets in the cytosol / positive regulation of cell motility / Regulation of TP53 Activity through Association with Co-factors / Co-inhibition by CTLA4 / Constitutive Signaling by AKT1 E17K in Cancer / negative regulation of PERK-mediated unfolded protein response / fat cell differentiation / Regulation of MITF-M-dependent genes involved in pigmentation / Regulation of localization of FOXO transcription factors / CD28 dependent PI3K/Akt signaling / Activation of BAD and translocation to mitochondria / positive regulation of protein targeting to membrane / Estrogen-dependent nuclear events downstream of ESR-membrane signaling / positive regulation of glycogen biosynthetic process / SARS-CoV-2 targets host intracellular signalling and regulatory pathways / Cyclin E associated events during G1/S transition / Cyclin A:Cdk2-associated events at S phase entry / Regulation of TP53 Activity through Acetylation / FLT3 Signaling / regulation of cell migration / molecular function activator activity / Downregulation of ERBB2:ERBB3 signaling / VEGFR2 mediated vascular permeability / positive regulation of D-glucose import across plasma membrane / protein localization to plasma membrane / TP53 Regulates Metabolic Genes / Translocation of SLC2A4 (GLUT4) to the plasma membrane / Deactivation of the beta-catenin transactivating complex / protein modification process / ruffle membrane / cellular response to insulin stimulus / glucose metabolic process / Regulation of PTEN stability and activity / insulin receptor signaling pathway / Regulation of TP53 Degradation / KEAP1-NFE2L2 pathway / G beta:gamma signalling through PI3Kgamma / PIP3 activates AKT signaling / cell cortex / early endosome / non-specific serine/threonine protein kinase / regulation of cell cycle / intracellular signal transduction / protein stabilization / positive regulation of cell migration / protein serine kinase activity / intracellular membrane-bounded organelle / protein serine/threonine kinase activity / signal transduction / nucleoplasm / ATP binding / metal ion binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function
Protein Kinase B beta, catalytic domain / Protein Kinase B, pleckstrin homology domain / Protein kinase, C-terminal / Protein kinase C terminal domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / Extension to Ser/Thr-type protein kinases / PH domain / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. ...Protein Kinase B beta, catalytic domain / Protein Kinase B, pleckstrin homology domain / Protein kinase, C-terminal / Protein kinase C terminal domain / Pleckstrin-homology domain (PH domain)/Phosphotyrosine-binding domain (PTB) / PH-domain like / Extension to Ser/Thr-type protein kinases / PH domain / AGC-kinase, C-terminal / AGC-kinase C-terminal domain profile. / PH domain profile. / Pleckstrin homology domain. / Pleckstrin homology domain / PH-like domain superfamily / Roll / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / Mainly Beta Similarity search - Domain/homology
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United Kingdom, 1items
United Kingdom, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads PDBj
PDBj





















 Assembly
Assembly
 Spodoptera (butterflies/moths) / References: UniProt: P31751
Spodoptera (butterflies/moths) / References: UniProt: P31751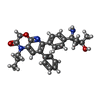




 Mass: 443.537 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H29N3O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 443.537 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H29N3O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: PO4 Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM
Mass: 195.237 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Sample preparation
Sample preparation / Beamline: X10SA / Wavelength: 1.00004 Å
/ Beamline: X10SA / Wavelength: 1.00004 Å Processing
Processing