 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8i70: Crystal structure of NADP-binding form of malonyl-CoA reductase C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8i70 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of NADP-binding form of malonyl-CoA reductase C-domain from Chloroflexus aurantiacus | ||||||
 Components Components | Short-chain dehydrogenase/reductase SDR | ||||||
 Keywords Keywords | OXIDOREDUCTASE / NAD(P)-binding protein / Short-chain reductase | ||||||
| Function / homology |  Function and homology information Function and homology informationfatty acid elongation / oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor / nucleotide binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Chloroflexus aurantiacus (bacteria) Chloroflexus aurantiacus (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Kim, S. / Kim, K.-J. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Int J Biol Macromol / Year: 2023 Title: Cryo-EM structure of bifunctional malonyl-CoA reductase from Chloroflexus aurantiacus reveals a dynamic domain movement for high enzymatic activity. Authors: Jae-Woo Ahn / Sangwoo Kim / Jiyeon Hong / Kyung-Jin Kim /  Abstract: The platform chemical 3-hydroxypropionic acid is used to synthesize various valuable materials, including bioplastics. Bifunctional malonyl-CoA reductase is a key enzyme in 3-hydroxypropionic acid ...The platform chemical 3-hydroxypropionic acid is used to synthesize various valuable materials, including bioplastics. Bifunctional malonyl-CoA reductase is a key enzyme in 3-hydroxypropionic acid biosynthesis as it catalyzes the two-step reduction of malonyl-CoA to malonate semialdehyde to 3-hydroxypropionic acid. Here, we report the cryo-EM structure of a full-length malonyl-CoA reductase protein from Chloroflexus aurantiacus (CaMCR). The EM model of CaMCR reveals a tandem helix architecture comprising an N-terminal (CaMCR) and a C-terminal (CaMCR) domain. The CaMCR model also revealed that the enzyme undergoes a dynamic domain movement between CaMCR and CaMCR due to the presence of a flexible linker between these two domains. Increasing the flexibility and extension of the linker resulted in a twofold increase in enzyme activity, indicating that for CaMCR, domain movement is crucial for high enzyme activity. We also describe the structural features of CaMCR and CaMCR. This study reveals the protein structures underlying the molecular mechanism of CaMCR and thereby provides valuable information for future enzyme engineering to improve the productivity of 3-hydroxypropionic acid. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8i70.cif.gz | 156.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8i70.ent.gz | 117.2 KB | Display | PDB format |
| PDBx/mmJSON format | 8i70.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i7/8i70ftp://data.pdbj.org/pub/pdb/validation_reports/i7/8i70 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8hjwC  8i6zSC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |

-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 75354.688 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Chloroflexus aurantiacus (bacteria) / Gene: Caur_2614 / Plasmid: pET28 / Production host: References: UniProt: A9WIU3, malonyl-CoA reductase (malonate semialdehyde-forming) | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-GOL /   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 | ||||||
| #3: Chemical | 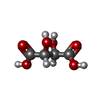  Mass: 150.087 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H6O6 / Feature type: SUBJECT OF INVESTIGATION Mass: 150.087 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H6O6 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | ChemComp-NAP / |   Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3 Mass: 743.405 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H28N7O17P3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 480 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.02 Å3/Da / Density % sol: 59.36 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, hanging drop / pH: 7 / Details: 0.1 M ammonium tartrate, 0.1 M Tris-Cl |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PAL/PLS / Beamline: 7A (6B, 6C1) / Wavelength: 0.97934 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 270 / Detector: CCD / Date: Nov 18, 2016 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Monochromator: DCM Si (111) Crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97934 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.9→50 Å / Num. obs: 67574 / % possible obs: 98.8 % / Redundancy: 2.1 % / Rmerge(I) obs: 0.064 / Χ2: 0.046 / Net I/σ(I): 12.9 / Num. measured all: 279616 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 8I6Z Resolution: 1.9→26.54 Å / Cor.coef. Fo:Fc: 0.969 / Cor.coef. Fo:Fc free: 0.951 / SU B: 2.322 / SU ML: 0.069 / Cross valid method: THROUGHOUT / ESU R: 0.109 / ESU R Free: 0.11 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.733 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 1.9→26.54 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|