National Institutes of Health/National Heart, Lung, and Blood Institute (NIH/NHLBI)
United States
Citation
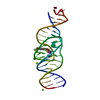
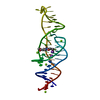
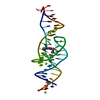
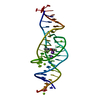
Journal: Nature / Year: 2023 Title: Intricate 3D architecture of a DNA mimic of GFP. Authors: Luiz F M Passalacqua / Michael T Banco / Jared D Moon / Xing Li / Samie R Jaffrey / Adrian R Ferré-D'Amaré / Abstract: Numerous studies have shown how RNA molecules can adopt elaborate three-dimensional (3D) architectures. By contrast, whether DNA can self-assemble into complex 3D folds capable of sophisticated ...Numerous studies have shown how RNA molecules can adopt elaborate three-dimensional (3D) architectures. By contrast, whether DNA can self-assemble into complex 3D folds capable of sophisticated biochemistry, independent of protein or RNA partners, has remained mysterious. Lettuce is an in vitro-evolved DNA molecule that binds and activates conditional fluorophores derived from GFP. To extend previous structural studies of fluorogenic RNAs, GFP and other fluorescent proteins to DNA, we characterize Lettuce-fluorophore complexes by X-ray crystallography and cryogenic electron microscopy. The results reveal that the 53-nucleotide DNA adopts a four-way junction (4WJ) fold. Instead of the canonical L-shaped or H-shaped structures commonly seen in 4WJ RNAs, the four stems of Lettuce form two coaxial stacks that pack co-linearly to form a central G-quadruplex in which the fluorophore binds. This fold is stabilized by stacking, extensive nucleobase hydrogen bonding-including through unusual diagonally stacked bases that bridge successive tiers of the main coaxial stacks of the DNA-and coordination of monovalent and divalent cations. Overall, the structure is more compact than many RNAs of comparable size. Lettuce demonstrates how DNA can form elaborate 3D structures without using RNA-like tertiary interactions and suggests that new principles of nucleic acid organization will be forthcoming from the analysis of complex DNAs.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



 PDBj
PDBj







































 Assembly
Assembly

 Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K
Mass: 39.098 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: K
 Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: Mg
 Mass: 281.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H9F2N3O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 281.215 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C12H9F2N3O3 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 16 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing