 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8dyp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of human cystine transporter cystinosin | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSPORT PROTEIN / Lysosomal transporter / Proton-coupled transporter / Cystine transporter / Cystinosis | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of melanin biosynthetic process / solute:proton symporter activity / L-cystine transmembrane transporter activity / L-cystine transport / : / Miscellaneous transport and binding events / regulation of TORC1 signaling / melanin biosynthetic process / grooming behavior / melanosome membrane ...regulation of melanin biosynthetic process / solute:proton symporter activity / L-cystine transmembrane transporter activity / L-cystine transport / : / Miscellaneous transport and binding events / regulation of TORC1 signaling / melanin biosynthetic process / grooming behavior / melanosome membrane / amino acid metabolic process / lens development in camera-type eye / adult walking behavior / long-term memory / ATP metabolic process / monoatomic ion transport / positive regulation of TORC1 signaling / glutathione metabolic process / brain development / visual learning / transmembrane transport / cognition / melanosome / late endosome / protein transport / lysosome / lysosomal membrane / extracellular exosome / plasma membrane Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å | ||||||
 Authors Authors | Guo, X. / Feng, L. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Cell / Year: 2022 Title: Structure and mechanism of human cystine exporter cystinosin. Authors: Xue Guo / Philip Schmiege / Tufa E Assafa / Rong Wang / Yan Xu / Linda Donnelly / Michael Fine / Xiaodan Ni / Jiansen Jiang / Glenn Millhauser / Liang Feng / Xiaochun Li / Abstract: Lysosomal amino acid efflux by proton-driven transporters is essential for lysosomal homeostasis, amino acid recycling, mTOR signaling, and maintaining lysosomal pH. To unravel the mechanisms of ...Lysosomal amino acid efflux by proton-driven transporters is essential for lysosomal homeostasis, amino acid recycling, mTOR signaling, and maintaining lysosomal pH. To unravel the mechanisms of these transporters, we focus on cystinosin, a prototypical lysosomal amino acid transporter that exports cystine to the cytosol, where its reduction to cysteine supplies this limiting amino acid for diverse fundamental processes and controlling nutrient adaptation. Cystinosin mutations cause cystinosis, a devastating lysosomal storage disease. Here, we present structures of human cystinosin in lumen-open, cytosol-open, and cystine-bound states, which uncover the cystine recognition mechanism and capture the key conformational states of the transport cycle. Our structures, along with functional studies and double electron-electron resonance spectroscopic investigations, reveal the molecular basis for the transporter's conformational transitions and protonation switch, show conformation-dependent Ragulator-Rag complex engagement, and demonstrate an unexpected activation mechanism. These findings provide molecular insights into lysosomal amino acid efflux and a potential therapeutic strategy. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8dyp.cif.gz | 101.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8dyp.ent.gz | 73.1 KB | Display | PDB format |
| PDBx/mmJSON format | 8dyp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/dy/8dypftp://data.pdbj.org/pub/pdb/validation_reports/dy/8dyp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  8dkeC  8dkiC  8dkmC  8dkwC  8dkxC  3p0gS  4x5nS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37173.371 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTNS / Production host: Homo sapiens (human) / References: UniProt: O60931 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Antibody | Mass: 13019.260 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host:  | ||||||
| #3: Chemical |   Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-OLC / ( | 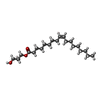  Mass: 356.540 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H40O4 Mass: 356.540 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H40O4Has ligand of interest | N | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.42 Å3/Da / Density % sol: 64.04 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: lipidic cubic phase / pH: 6.5 / Details: 12% PEG400, 80mM Li2SO4, and 50mM MES pH6.0 / PH range: 6.0-6.8 |
-Data collection
| Diffraction |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||
| Detector |
| ||||||||||||||||||||
| Radiation |
| ||||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||||
| Reflection | Resolution: 3.4→40.19 Å / Num. obs: 9815 / % possible obs: 97.3 % / Redundancy: 14.9 % / Biso Wilson estimate: 105.72 Å2 / CC1/2: 0.967 / CC star: 0.992 / Net I/σ(I): 15.2 | ||||||||||||||||||||
| Reflection shell | Resolution: 3.4→3.52 Å / Redundancy: 8.3 % / Mean I/σ(I) obs: 1.69 / Num. unique obs: 886 / CC1/2: 0.722 / % possible all: 88.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3P0G, 4x5n Resolution: 3.4→40.19 Å / SU ML: 0.4795 / Cross valid method: FREE R-VALUE / Phase error: 34.0292 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 100.5 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.4→40.19 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
