 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8c41: High resolution structure of the Streptococcus pneumoniae topoiso... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8c41 | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution structure of the Streptococcus pneumoniae topoisomerase IV-DNA complex with the novel fluoroquinolone Delafloxacin | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ISOMERASE / PROTEIN-DNA CLEAVAGE COMPLEX / TOPOISOMERASE IIA / DELAFLOXACIN / TOPOISOMERASE IV-DNA-ANTIBIOTIC COMPLEX | ||||||
| Function / homology | delafloxacin / DNA / DNA (> 10) Function and homology information Function and homology information | ||||||
| Biological species |   Streptococcus pneumoniae (bacteria) Streptococcus pneumoniae (bacteria)DNA molecule (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.393 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.393 Å | ||||||
 Authors Authors | Najmudin, S. / Pan, X.S. / Wang, B. / Chayen, N.E. / Fisher, L.M. / Sanderson, M.R. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2025 Title: Structural basis of topoisomerase targeting by delafloxacin. Authors: Najmudin, S. / Pan, X.S. / Wang, B. / Govada, L. / Chayen, N.E. / Rubio, N. / Shaffer, M.S.P. / Rzepa, H.S. / Fisher, L.M. / Sanderson, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8c41.cif.gz | 1.5 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8c41.ent.gz | 963.5 KB | Display | PDB format |
| PDBx/mmJSON format | 8c41.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c4/8c41ftp://data.pdbj.org/pub/pdb/validation_reports/c4/8c41 | HTTPS FTP |
|---|
-Related structure data


-Links
 PDBj
PDBj






































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 84211.422 Da / Num. of mol.: 2 / Mutation: Insertion of His at postion 648 Source method: isolated from a genetically manipulated source Details: The fused TOPOISOMERASE IV CLEAVAGE COMPLEX comprises the C-terminal domain of the ParE30 domain (residues 415-647), a His insert at position 648 and the N-terminal domain of ParC55 (residues 1001-1486) Source: (gene. exp.) Streptococcus pneumoniae (bacteria) / Strain: 7785 / Gene: SP_0852-SP_0855 / Plasmid: PET29A / Production host: References: Isomerases; Other isomerases; Sole sub-subclass for isomerases that do not belong in the other subclasses |
|---|
-DNA chain , 4 types, 4 molecules EFGH
| #2: DNA chain | Mass: 2121.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) DNA molecule (others) |
|---|---|
| #3: DNA chain | Mass: 3348.209 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: cleaved from the E18 mer oligo AGTCATTCATGACCTTGGT / Source: (synth.) DNA molecule (others) |
| #4: DNA chain | Mass: 2113.410 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) DNA molecule (others) |
| #5: DNA chain | Mass: 3358.211 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) DNA molecule (others) |
-Non-polymers , 5 types, 431 molecules 


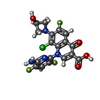

| #6: Chemical |  Mass: 118.174 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#7: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION#8: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#9: Chemical |  Mass: 440.760 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H12ClF3N4O4 / Feature type: SUBJECT OF INVESTIGATION / Comment: antibiotic*YM Mass: 440.760 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H12ClF3N4O4 / Feature type: SUBJECT OF INVESTIGATION / Comment: antibiotic*YM#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 418 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.24 Å3/Da / Density % sol: 71 % |
|---|---|
| Crystal grow | Temperature: 300 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 2.5% Tacsimate, 50 mM Na Cacodylate, 62.5 mM KCl, 7.5 mM MgCl2, 4-7.5% Isopropanol. 30% MPD as cryoprotectant PH range: 6.5-8.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04 / Wavelength: 0.95373 Å |
| Detector | Type: DECTRIS EIGER2 XE 16M / Detector: PIXEL / Date: Oct 15, 2022 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95373 Å / Relative weight: 1 |
| Reflection | Resolution: 2.393→115.01 Å / Num. obs: 70836 / % possible obs: 96.3 % / Observed criterion σ(I): 2 / Redundancy: 60.8 % / CC1/2: 1 / Rmerge(I) obs: 0.564 / Rpim(I) all: 0.073 / Rrim(I) all: 0.569 / Net I/σ(I): 8.4 |
| Reflection shell | Resolution: 2.394→2.689 Å / Redundancy: 49.5 % / Rmerge(I) obs: 4.363 / Mean I/σ(I) obs: 1.8 / Num. unique obs: 3517 / CC1/2: 0.83 / Rpim(I) all: 0.62 / Rrim(I) all: 4.483 / % possible all: 84 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.393→115.01 Å / Cor.coef. Fo:Fc: 0.932 / Cor.coef. Fo:Fc free: 0.899 / WRfactor Rfree: 0.219 / WRfactor Rwork: 0.175 / SU B: 14.946 / SU ML: 0.187 / Average fsc free: 0.9534 / Average fsc work: 0.9697 / Cross valid method: FREE R-VALUE / ESU R: 0.492 / ESU R Free: 0.307 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.207 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.393→115.01 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
