 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8qmc: High resolution structure of the Streptococcus pneumoniae topoiso... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8qmc | ||||||
|---|---|---|---|---|---|---|---|
| Title | High resolution structure of the Streptococcus pneumoniae topoisomerase IV-complex with the V-site 18mer dsDNA and novel fluoroquinolone Delafloxacin | ||||||
 Components Components |
| ||||||
 Keywords Keywords | ISOMERASE / PROTEIN-DNA CLEAVAGE COMPLEX / TOPOISOMERASE IIA / DELAFLOXACIN / TOPOISOMERASE IV-DNA-ANTIBIOTIC COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationDNA topoisomerase type II (double strand cut, ATP-hydrolyzing) complex / DNA negative supercoiling activity / DNA topoisomerase (ATP-hydrolysing) / extrinsic component of plasma membrane / DNA topological change / chromosome segregation / chromosome / DNA binding / ATP binding / cytoplasm Similarity search - Function | ||||||
| Biological species |   Streptococcus pneumoniae (bacteria) Streptococcus pneumoniae (bacteria)DNA molecule (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.402 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.402 Å | ||||||
 Authors Authors | Najmudin, S. / Pan, X.S. / Wang, B. / Chayen, N.E. / Fisher, L.M. / Sanderson, M.R. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2025 Title: Structural basis of topoisomerase targeting by delafloxacin. Authors: Najmudin, S. / Pan, X.S. / Wang, B. / Govada, L. / Chayen, N.E. / Rubio, N. / Shaffer, M.S.P. / Rzepa, H.S. / Fisher, L.M. / Sanderson, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8qmc.cif.gz | 1.5 MB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8qmc.ent.gz | 975.5 KB | Display | PDB format |
| PDBx/mmJSON format | 8qmc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qm/8qmcftp://data.pdbj.org/pub/pdb/validation_reports/qm/8qmc | HTTPS FTP |
|---|
-Related structure data


-Links
 PDBj
PDBj







































- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 84211.422 Da / Num. of mol.: 2 / Mutation: Insertion of His at postion 648 Source method: isolated from a genetically manipulated source Details: he fused TOPOISOMERASE IV CLEAVAGE COMPLEX comprises the C-terminal domain of the ParE30 domain (residues 415-647), a His insert at position 648 and the N-terminal domain of ParC55 (residues ...Details: he fused TOPOISOMERASE IV CLEAVAGE COMPLEX comprises the C-terminal domain of the ParE30 domain (residues 415-647), a His insert at position 648 and the N-terminal domain of ParC55 (residues 1001-1486),he fused TOPOISOMERASE IV CLEAVAGE COMPLEX comprises the C-terminal domain of the ParE30 domain (residues 415-647), a His insert at position 648 and the N-terminal domain of ParC55 (residues 1001-1486) Source: (gene. exp.) Streptococcus pneumoniae (bacteria) / Strain: 7785 / Gene: AMCSP13_000989, parC, SP_0855 / Plasmid: pET29A / Production host: References: UniProt: J0V1V8, UniProt: P72525, DNA topoisomerase (ATP-hydrolysing) |
|---|
-DNA chain , 4 types, 4 molecules EFHG
| #2: DNA chain | Mass: 2168.445 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: pBR322 / Source: (synth.) DNA molecule (others) |
|---|---|
| #3: DNA chain | Mass: 3333.198 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: pBR322 / Source: (synth.) DNA molecule (others) |
| #4: DNA chain | Mass: 3317.199 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: pBR322 / Source: (synth.) DNA molecule (others) |
| #5: DNA chain | Mass: 2121.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Details: pBR322 / Source: (synth.) DNA molecule (others) |
-Non-polymers , 7 types, 441 molecules 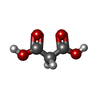




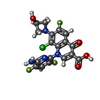

| #6: Chemical |  Mass: 104.061 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O4 Mass: 104.061 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H4O4#7: Chemical |  Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2#8: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Mg#9: Chemical |  Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Cl#10: Chemical | ChemComp-MPD / ( |  Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM Mass: 118.174 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM#11: Chemical |  Mass: 440.760 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H12ClF3N4O4 / Feature type: SUBJECT OF INVESTIGATION / Comment: antibiotic*YM Mass: 440.760 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C18H12ClF3N4O4 / Feature type: SUBJECT OF INVESTIGATION / Comment: antibiotic*YM#12: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 426 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.21 Å3/Da / Density % sol: 71.1 % |
|---|---|
| Crystal grow | Temperature: 301 K / Method: vapor diffusion, sitting drop Details: 2.5% Tacsimate, 50 mM Na Cacodylate, 62.5 mM KCl, 7.5 mM MgCl2, 5.5-7.0% Isopropanol. 30% MPD as cryoprotectant |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04 / Wavelength: 0.95373 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Mar 9, 2023 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.95373 Å / Relative weight: 1 |
| Reflection | Resolution: 2.402→64.862 Å / Num. obs: 60189 / % possible obs: 96.2 % / Redundancy: 20.1 % / CC1/2: 0.997 / Rmerge(I) obs: 0.341 / Rpim(I) all: 0.078 / Rrim(I) all: 0.349 / Net I/σ(I): 10.3 |
| Reflection shell | Resolution: 2.402→2.891 Å / Redundancy: 20.8 % / Rmerge(I) obs: 2.05 / Mean I/σ(I) obs: 2 / Num. unique obs: 5142 / CC1/2: 0.745 / Rpim(I) all: 0.459 / Rrim(I) all: 2.102 / % possible all: 86.2 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.402→64.862 Å / Cor.coef. Fo:Fc: 0.873 / Cor.coef. Fo:Fc free: 0.814 / SU B: 23.757 / SU ML: 0.265 / Cross valid method: FREE R-VALUE / ESU R: 1.101 / ESU R Free: 0.434 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 26.352 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.402→64.862 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection: ALL |
