 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-8afr: Pim1 in complex with 4-((6-hydroxybenzofuran-3-yl)methyl)benzoic ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 8afr | ||||||
|---|---|---|---|---|---|---|---|
| Title | Pim1 in complex with 4-((6-hydroxybenzofuran-3-yl)methyl)benzoic acid and Pimtide | ||||||
 Components Components |
| ||||||
 Keywords Keywords | TRANSFERASE / SERINE KINASE / KINASE / COMPLEX / PIM1 / PIM / PIM-1 / INHIBITOR / TUMORIGENISIS / CANCER / PIMTIDE / PROTO ONCOGEN / ATP / PHOSPHORYLATION / APOPTOSIS / CELL CYCLE | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / positive regulation of protein serine/threonine kinase activity / ribosomal small subunit binding ...regulation of transmembrane transporter activity / positive regulation of cardioblast proliferation / positive regulation of cyclin-dependent protein serine/threonine kinase activity / regulation of hematopoietic stem cell proliferation / vitamin D receptor signaling pathway / cellular detoxification / STAT5 activation downstream of FLT3 ITD mutants / transcription factor binding / positive regulation of protein serine/threonine kinase activity / ribosomal small subunit binding / : / positive regulation of cardiac muscle cell proliferation / positive regulation of brown fat cell differentiation / Signaling by FLT3 fusion proteins / positive regulation of TORC1 signaling / regulation of mitotic cell cycle / negative regulation of innate immune response / protein serine/threonine kinase activator activity / cellular response to type II interferon / protein autophosphorylation / manganese ion binding / Interleukin-4 and Interleukin-13 signaling / protein phosphorylation / non-specific serine/threonine protein kinase / protein stabilization / protein serine kinase activity / protein serine/threonine kinase activity / apoptotic process / negative regulation of apoptotic process / positive regulation of DNA-templated transcription / nucleolus / nucleoplasm / ATP binding / nucleus / plasma membrane / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)Synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å | ||||||
 Authors Authors | Hochban, P.M.M. / Heine, A. / Diederich, W.E. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Eur.J.Med.Chem. / Year: 2023 Title: Pose, duplicate, then elaborate: Steps towards increased affinity for inhibitors targeting the specificity surface of the Pim-1 kinase. Authors: Heyder, L. / Hochban, P.M.M. / Taylor, C. / Chevillard, F. / Siefker, C. / Iking, C. / Borchardt, H. / Aigner, A. / Klebe, G. / Heine, A. / Kolb, P. / Diederich, W.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 8afr.cif.gz | 153.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb8afr.ent.gz | 99 KB | Display | PDB format |
| PDBx/mmJSON format | 8afr.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/af/8afrftp://data.pdbj.org/pub/pdb/validation_reports/af/8afr | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7qb2C  7qfmC  7z6uC  5ndtS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 35356.141 Da / Num. of mol.: 1 / Mutation: R250G Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PIM1 / Plasmid: pLIC-SGC / Production host:  References: UniProt: P11309, non-specific serine/threonine protein kinase |
|---|---|
| #2: Protein/peptide | Mass: 1592.850 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) Synthetic construct (others) |
| #3: Chemical | ChemComp-M0F / 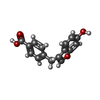  Mass: 268.264 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H12O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 268.264 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H12O4 / Feature type: SUBJECT OF INVESTIGATION |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 111 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3 Å3/Da / Density % sol: 59.4 % |
|---|---|
| Crystal grow | Temperature: 291.15 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 100 mM bis-tris-propane (pH7.0), 10 % ethylene glycol, 0.3% DMSO, 20% PEG3350, 200 mM LiCl PH range: 6.8 - 7.2 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 7, 2019 / Details: Sagitally bended Si111-crystal |
| Radiation | Monochromator: Double crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2.15→48.22 Å / Num. obs: 22836 / % possible obs: 98.1 % / Redundancy: 13.9 % / Biso Wilson estimate: 40.22 Å2 / CC1/2: 1 / Rsym value: 0.055 / Net I/σ(I): 32.97 |
| Reflection shell | Resolution: 2.15→2.28 Å / Redundancy: 13.9 % / Mean I/σ(I) obs: 5.12 / Num. unique obs: 3583 / CC1/2: 0.963 / % possible all: 95.7 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5NDT Resolution: 2.15→48.22 Å / SU ML: 0.1586 / Cross valid method: FREE R-VALUE / σ(F): 1.36 / Phase error: 19.9775 / Stereochemistry target values: GeoStd + Monomer Library
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 49.77 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.15→48.22 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
