- PDB-7t9x: Saccharomyces cerevisiae Pex12 RING domain -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 7t9x
Title
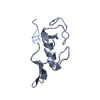
Saccharomyces cerevisiae Pex12 RING domain
Components
Peroxisome assembly protein 12
Keywords
LIGASE / Saccharomyces cerevisiae Pex12 RING domain
Function / homology
Function and homology information
protein import into peroxisome matrix, substrate release / : / peroxisomal importomer complex / protein import into peroxisome matrix, receptor recycling / Class I peroxisomal membrane protein import / protein import into peroxisome matrix / Peroxisomal protein import / ubiquitin ligase activator activity / peroxisomal membrane / transmembrane protein transporter activity ...protein import into peroxisome matrix, substrate release / : / peroxisomal importomer complex / protein import into peroxisome matrix, receptor recycling / Class I peroxisomal membrane protein import / protein import into peroxisome matrix / Peroxisomal protein import / ubiquitin ligase activator activity / peroxisomal membrane / transmembrane protein transporter activity / protein monoubiquitination / ubiquitin ligase complex / protein polyubiquitination / ubiquitin-protein transferase activity / ubiquitin protein ligase activity / proteasome-mediated ubiquitin-dependent protein catabolic process / protein ubiquitination / zinc ion binding Similarity search - Function
Journal: Nature / Year: 2022 Title: A peroxisomal ubiquitin ligase complex forms a retrotranslocation channel. Authors: Peiqiang Feng / Xudong Wu / Satchal K Erramilli / Joao A Paulo / Pawel Knejski / Steven P Gygi / Anthony A Kossiakoff / Tom A Rapoport / Abstract: Peroxisomes are ubiquitous organelles that house various metabolic reactions and are essential for human health. Luminal peroxisomal proteins are imported from the cytosol by mobile receptors, which ...Peroxisomes are ubiquitous organelles that house various metabolic reactions and are essential for human health. Luminal peroxisomal proteins are imported from the cytosol by mobile receptors, which then recycle back to the cytosol by a poorly understood process. Recycling requires receptor modification by a membrane-embedded ubiquitin ligase complex comprising three RING finger domain-containing proteins (Pex2, Pex10 and Pex12). Here we report a cryo-electron microscopy structure of the ligase complex, which together with biochemical and in vivo experiments reveals its function as a retrotranslocation channel for peroxisomal import receptors. Each subunit of the complex contributes five transmembrane segments that co-assemble into an open channel. The three ring finger domains form a cytosolic tower, with ring finger 2 (RF2) positioned above the channel pore. We propose that the N terminus of a recycling receptor is inserted from the peroxisomal lumen into the pore and monoubiquitylated by RF2 to enable extraction into the cytosol. If recycling is compromised, receptors are polyubiquitylated by the concerted action of RF10 and RF12 and degraded. This polyubiquitylation pathway also maintains the homeostasis of other peroxisomal import factors. Our results clarify a crucial step during peroxisomal protein import and reveal why mutations in the ligase complex cause human disease.
Component-ID: 1 / Ens-ID: 1 / Beg auth comp-ID: PRO / Beg label comp-ID: PRO / End auth comp-ID: ILE / End label comp-ID: ILE / Auth seq-ID: 2 - 74 / Label seq-ID: 1 - 73
Dom-ID
Selection details
Auth asym-ID
Label asym-ID
1
chain 'A'
A
A
2
chain 'B'
B
B
-
Components
#1: Protein
Peroxisomeassemblyprotein12 / Peroxin-12
Mass: 8008.321 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Bacteria (eubacteria) / Gene: PEX12, YMR026C, YM9711.16C / Production host: Bacteria (eubacteria) / References: UniProt: Q04370
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads

 PDBj
PDBj

 Assembly
Assembly


 Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION
Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 74 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing