 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7rz1: Hen egg-white lysozyme with ionic liquid ethanolammonium formate ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7rz1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Hen egg-white lysozyme with ionic liquid ethanolammonium formate 14.4 mol% | |||||||||
 Components Components | Lysozyme C | |||||||||
 Keywords Keywords | HYDROLASE / lysozyme / ionic liquid / ethanolammonium formate | |||||||||
| Function / homology |  Function and homology information Function and homology informationLactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium ...Lactose synthesis / Antimicrobial peptides / Neutrophil degranulation / beta-N-acetylglucosaminidase activity / cell wall macromolecule catabolic process / lysozyme / lysozyme activity / killing of cells of another organism / defense response to Gram-negative bacterium / defense response to bacterium / defense response to Gram-positive bacterium / endoplasmic reticulum / : / identical protein binding / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.046 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.046 Å | |||||||||
 Authors Authors | Han, Q. / Darmanin, C. / Drummond, C. / Greaves, T. | |||||||||
| Funding support |  Australia, 1items Australia, 1items
| |||||||||
 Citation Citation | Journal: J Colloid Interface Sci / Year: 2023 Title: Probing ion-binding at a protein interface: Modulation of protein properties by ionic liquids. Authors: Han, Q. / Su, Y. / Smith, K.M. / Binns, J. / Drummond, C.J. / Darmanin, C. / Greaves, T.L. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7rz1.cif.gz | 114.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7rz1.ent.gz | 84.8 KB | Display | PDB format |
| PDBx/mmJSON format | 7rz1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/rz/7rz1ftp://data.pdbj.org/pub/pdb/validation_reports/rz/7rz1 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7rxyC  7rydC  7rykC  7rz0C  7rz2C  7jmuS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 14331.160 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-FMT /   Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 46.025 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: CH2O2 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | 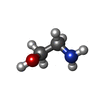  Type: L-peptide COOH carboxy terminus / Mass: 61.083 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H7NO / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide COOH carboxy terminus / Mass: 61.083 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H7NO / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 106 / Source method: isolated from a natural source / Formula: H2OCompound details | The protein is a monomer, but is an oligomer in the presence of concentrated ionic liquid. Under ...The protein is a monomer, but is an oligomer in the presence of concentrated ionic liquid. Under these conditions, SAXS indicates that the radius of gyration increased to over 20 angstroms compared with 15 angstroms in buffer. | Has ligand of interest | Y | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.96 Å3/Da / Density % sol: 37.31 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, hanging drop Details: ethanolammonium formate 14.4mol% (ca. 50wt%) in water |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Australian Synchrotron / Beamline: MX1 / Wavelength: 1.2398 Å |
| Detector | Type: ADSC QUANTUM 210r / Detector: CCD / Date: May 2, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.2398 Å / Relative weight: 1 |
| Reflection | Resolution: 2.046→54.2 Å / Num. obs: 7613 / % possible obs: 100 % / Redundancy: 10.7 % / CC1/2: 0.981 / Net I/σ(I): 11.3 |
| Reflection shell | Resolution: 2.05→2.1 Å / Mean I/σ(I) obs: 2.8 / Num. unique obs: 571 / CC1/2: 0.852 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 7JMU Resolution: 2.046→54.2 Å / Cor.coef. Fo:Fc: 0.957 / Cor.coef. Fo:Fc free: 0.907 / WRfactor Rfree: 0.21 / WRfactor Rwork: 0.14 / SU B: 13.824 / SU ML: 0.156 / Average fsc free: 0.9114 / Average fsc work: 0.9306 / Cross valid method: THROUGHOUT / ESU R Free: 0.207 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 18.825 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.046→54.2 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: -19.5967 Å / Origin y: -1.6795 Å / Origin z: -9.3171 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection: ALL |
