 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7r3g: PROSS optimitzed variant of RhlR (75 mutations) in complex with t... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7r3g | ||||||
|---|---|---|---|---|---|---|---|
| Title | PROSS optimitzed variant of RhlR (75 mutations) in complex with the synthetic antagonist mBTL | ||||||
 Components Components | Regulatory protein RhlR | ||||||
 Keywords Keywords | TRANSCRIPTION / QUORUM SENSING / LuxR-type TRANSCRIPTIONAL REGULATOR / PSEUDOMONAS / DNA BINDING PROTEIN / GENE REGULATION / mBTL / rhamnolipid | ||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of pyocyanine biosynthetic process / positive regulation of secondary metabolite biosynthetic process / quorum sensing / DNA-binding transcription repressor activity / DNA-binding transcription activator activity / positive regulation of proteolysis / positive regulation of lipid biosynthetic process / protein-DNA complex / signaling receptor activity / gene expression ...positive regulation of pyocyanine biosynthetic process / positive regulation of secondary metabolite biosynthetic process / quorum sensing / DNA-binding transcription repressor activity / DNA-binding transcription activator activity / positive regulation of proteolysis / positive regulation of lipid biosynthetic process / protein-DNA complex / signaling receptor activity / gene expression / transcription cis-regulatory region binding / DNA-binding transcription factor activity / positive regulation of DNA-templated transcription / cytoplasm Similarity search - Function | ||||||
| Biological species |  Pseudomonas aeruginosa PAO1 (bacteria) Pseudomonas aeruginosa PAO1 (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.15 Å | ||||||
 Authors Authors | Henke, S. / Blankenfeldt, W. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2022 Title: Moonlighting chaperone activity of the enzyme PqsE contributes to RhlR-controlled virulence of Pseudomonas aeruginosa. Authors: Borgert, S.R. / Henke, S. / Witzgall, F. / Schmelz, S. / Zur Lage, S. / Hotop, S.K. / Stephen, S. / Lubken, D. / Kruger, J. / Gomez, N.O. / van Ham, M. / Jansch, L. / Kalesse, M. / Pich, A. ...Authors: Borgert, S.R. / Henke, S. / Witzgall, F. / Schmelz, S. / Zur Lage, S. / Hotop, S.K. / Stephen, S. / Lubken, D. / Kruger, J. / Gomez, N.O. / van Ham, M. / Jansch, L. / Kalesse, M. / Pich, A. / Bronstrup, M. / Haussler, S. / Blankenfeldt, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7r3g.cif.gz | 286.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7r3g.ent.gz | 238 KB | Display | PDB format |
| PDBx/mmJSON format | 7r3g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/r3/7r3gftp://data.pdbj.org/pub/pdb/validation_reports/r3/7r3g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7r3eC  7r3fC  7r3hC  7r3iC  7r3jC  8b4aC C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27797.574 Da / Num. of mol.: 2 Mutation: L9D: D12E: G13D: P20S: H22T: G26E: G46C: T50P: K57R: T58I: E59F: V60M: H61F: T63N: K66P: L69Q: R71H: M74A: G78F: V80I: A83T: L85R: N86H: G87C: S91G: E92N: M93H: V94I: S99D: D102A: Q103D: ...Mutation: L9D: D12E: G13D: P20S: H22T: G26E: G46C: T50P: K57R: T58I: E59F: V60M: H61F: T63N: K66P: L69Q: R71H: M74A: G78F: V80I: A83T: L85R: N86H: G87C: S91G: E92N: M93H: V94I: S99D: D102A: Q103D: S104A: R105Q: M106E: N109D: E110D: W114Y: C117R: V118H: L122H: P123S: I124C: R125M: N129G: L130V: L131M: S132G: V133F: D139S: Q140S: Q141P: N142A: S145P: F146H: I151L: L155M: M158L: T163H: K165T: D168E: E170N: M173S: M175Q: S176P: N177Q: V179I: H183K: Q190R: S197T: G199A: I203K: S206G: H216L: D224N: L230Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudomonas aeruginosa PAO1 (bacteria)Strain: ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1 Gene: rhlR, lasM, vsmR, PA3477 / Production host: #2: Chemical | 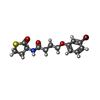  Mass: 358.251 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H16BrNO3S / Feature type: SUBJECT OF INVESTIGATION Mass: 358.251 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H16BrNO3S / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 136 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 136 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 57.8 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 0.1 M Bis-Tris pH 6.5, 0.2 M NaCl, 25 % (w/v) PEG 3350 3.5 mg/ml protein cryoprotectant: 10 % (v/v) (2R,3R) -(-)-2,3-Butanediol |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, DESY / Beamline: P11 / Wavelength: 1.0332 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Mar 13, 2020 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1.0332 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.15→47.57 Å / Num. obs: 34628 / % possible obs: 100 % / Redundancy: 20.1 % / Biso Wilson estimate: 35.03 Å2 / CC1/2: 0.998 / Rmerge(I) obs: 0.231 / Rpim(I) all: 0.053 / Rrim(I) all: 0.238 / Net I/σ(I): 11.1 / Num. measured all: 697687 / Scaling rejects: 1 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: structure prediction of phyre2 Resolution: 2.15→47.57 Å / SU ML: 0.24 / Cross valid method: THROUGHOUT / σ(F): 1.33 / Phase error: 24.45 / Stereochemistry target values: ML
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 141.8 Å2 / Biso mean: 51.2426 Å2 / Biso min: 21.46 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.15→47.57 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 12 / % reflection obs: 100 %
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
