 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7q1f | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CPAP:TUBULIN:IE5 ALPHAREP COMPLEX | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | CELL CYCLE / MICROTUBULE / CENTROMERE PROTEIN J / CENTRIOLE | |||||||||
| Function / homology |  Function and homology information Function and homology informationmicrotubule-based process / structural constituent of cytoskeleton / microtubule / GTP binding / cytoplasm Similarity search - Function | |||||||||
| Biological species | synthetic construct (others) Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.347 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.347 Å | |||||||||
 Authors Authors | Campanacci, V. / Gigant, b. | |||||||||
| Funding support |  France, 2items France, 2items
| |||||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 2022 Title: Structural convergence for tubulin binding of CPAP and vinca domain microtubule inhibitors. Authors: Campanacci, V. / Urvoas, A. / Ammar Khodja, L. / Aumont-Nicaise, M. / Noiray, M. / Lachkar, S. / Curmi, P.A. / Minard, P. / Gigant, B. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7q1f.cif.gz | 1020.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7q1f.ent.gz | 833.8 KB | Display | PDB format |
| PDBx/mmJSON format | 7q1f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/q1/7q1fftp://data.pdbj.org/pub/pdb/validation_reports/q1/7q1f | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7q1eC  7z0fC  7z0gC  6gwcS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj








- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 4 types, 10 molecules ARBSCDTUPV
| #1: Protein | Mass: 50204.445 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) #2: Protein | Mass: 49999.887 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) #3: Protein | Mass: 25173.736 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) synthetic construct (others) / Production host:  #4: Protein | Mass: 8606.816 Da / Num. of mol.: 2 Mutation: V319M, A320V, A361del, E362del, P364G, L365G, P366G, I367G, K368del, A369del Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CENPJ, CPAP, LAP, LIP1 / Production host: |
|---|
-Non-polymers , 7 types, 725 molecules 




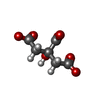

| #5: Chemical |  Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM Mass: 523.180 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16N5O14P3 / Comment: GTP, energy-carrying molecule*YM#6: Chemical |  Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg#7: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#8: Chemical |  Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM Type: RNA linking / Mass: 443.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O11P2 / Comment: GDP, energy-carrying molecule*YM#9: Chemical | ChemComp-MES /  Mass: 195.237 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#10: Chemical | ChemComp-FLC / |  Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H5O7#11: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 712 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.72 Å3/Da / Density % sol: 54.75 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion / pH: 6.8 Details: 0.18 M tri-ammonium citrate, 0.1 M Mes-K pH 6.8, 18.5 % PEG3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL / Beamline: PROXIMA 1 / Wavelength: 0.97856 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Mar 19, 2021 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97856 Å / Relative weight: 1 |
| Reflection | Resolution: 2.347→107.932 Å / Num. obs: 97303 / % possible obs: 90.8 % / Redundancy: 7.2 % / CC1/2: 0.994 / Rpim(I) all: 0.07 / Rrim(I) all: 0.189 / Net I/σ(I): 7.1 |
| Reflection shell | Resolution: 2.347→2.609 Å / Mean I/σ(I) obs: 1.6 / Num. unique obs: 4866 / CC1/2: 0.675 / Rpim(I) all: 0.415 / Rrim(I) all: 1.107 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6GWC Resolution: 2.347→107.93 Å / Cor.coef. Fo:Fc: 0.939 / Cor.coef. Fo:Fc free: 0.921 / SU R Cruickshank DPI: 0.562 / Cross valid method: THROUGHOUT / SU R Blow DPI: 0.526 / SU Rfree Blow DPI: 0.26 / SU Rfree Cruickshank DPI: 0.266
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 65.63 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.31 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.347→107.93 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.35→2.51 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
