 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7p7n: X-RAY CRYSTAL STRUCTURE OF SPOROSARCINA PASTEURII UREASE INHIBITE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7p7n | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-RAY CRYSTAL STRUCTURE OF SPOROSARCINA PASTEURII UREASE INHIBITED BY THE GOLD(I)-PHOSPHINE COMPOUND Au(PEt3)I DETERMINED AT 1.80 ANGSTROMS | ||||||
 Components Components | (Urease subunit ...) x 3 | ||||||
 Keywords Keywords | HYDROLASE / urease / nickel / gold / enzyme / urea | ||||||
| Function / homology |  Function and homology information Function and homology informationurease complex / urease / urease activity / urea catabolic process / nickel cation binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  Sporosarcina pasteurii (bacteria) Sporosarcina pasteurii (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Mazzei, L. / Ciurli, S. | ||||||
| Funding support | 1items
| ||||||
 Citation Citation | Journal: Dalton Trans / Year: 2021 Title: Medicinal Au(I) compounds targeting urease as prospective antimicrobial agents: unveiling the structural basis for enzyme inhibition. Authors: Mazzei, L. / Massai, L. / Cianci, M. / Messori, L. / Ciurli, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7p7n.cif.gz | 195.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7p7n.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 7p7n.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p7/7p7nftp://data.pdbj.org/pub/pdb/validation_reports/p7/7p7n | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7p7oC  5ol4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

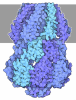



- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||
| Components on special symmetry positions |
|
-Components
-Urease subunit ... , 3 types, 3 molecules AAABBBCCC
| #1: Protein | Mass: 11134.895 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Sporosarcina pasteurii (bacteria) / References: UniProt: P41022, urease |
|---|---|
| #2: Protein | Mass: 13529.061 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Sporosarcina pasteurii (bacteria) / References: UniProt: P41021, urease |
| #3: Protein | Mass: 61575.648 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Sporosarcina pasteurii (bacteria) / References: UniProt: P41020, urease |
-Non-polymers , 7 types, 585 molecules 



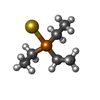


| #4: Chemical | ChemComp-EDO /  Mass: 62.068 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C2H6O2#5: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 15 / Source method: obtained synthetically / Formula: SO4#6: Chemical |  Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ni#7: Chemical | ChemComp-O / |  Mass: 15.999 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: O Mass: 15.999 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: O#8: Chemical | ChemComp-AUF / |  Mass: 315.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15AuP Mass: 315.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H15AuP#9: Chemical | ChemComp-AU /  Mass: 196.967 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Au Mass: 196.967 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Au#10: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 548 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | N |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.77 Å3/Da / Density % sol: 55.6 % / Description: rice shaped |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: THE PROTEIN-LIGAND (1.2 mM) COMPLEX IN 50 MM HEPES BUFFER, PH 7.50 (ALSO CONTAINING 10% (V/V) DMSO), DILUTED 1:1 WITH A SOLUTION OF 1.2-1.7 M AMMONIUM SULFATE ALSO CONTAINING THE SAME ...Details: THE PROTEIN-LIGAND (1.2 mM) COMPLEX IN 50 MM HEPES BUFFER, PH 7.50 (ALSO CONTAINING 10% (V/V) DMSO), DILUTED 1:1 WITH A SOLUTION OF 1.2-1.7 M AMMONIUM SULFATE ALSO CONTAINING THE SAME CONCENTRATION OF LIGAND AND DMSO. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY  / Beamline: P13 (MX1) / Wavelength: 0.9762 Å / Beamline: P13 (MX1) / Wavelength: 0.9762 Å |
| Detector | Type: DECTRIS PILATUS 6M-F / Detector: PIXEL / Date: Nov 30, 2019 |
| Radiation | Monochromator: Si(III) crystal / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9762 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→114 Å / Num. obs: 89759 / % possible obs: 100 % / Redundancy: 13.2 % / Biso Wilson estimate: 25.26 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.142 / Rpim(I) all: 0.042 / Rrim(I) all: 0.154 / Net I/σ(I): 17 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 13.5 % / Rmerge(I) obs: 2.3 / Mean I/σ(I) obs: 1.5 / Num. unique obs: 4534 / CC1/2: 0.694 / Rpim(I) all: 0.931 / Rrim(I) all: 2.484 / % possible all: 100 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5ol4 Resolution: 1.8→72.907 Å / Cor.coef. Fo:Fc: 0.975 / Cor.coef. Fo:Fc free: 0.97 / WRfactor Rfree: 0.15 / WRfactor Rwork: 0.131 / SU B: 2.666 / SU ML: 0.075 / Average fsc free: 0.918 / Average fsc work: 0.9231 / Cross valid method: FREE R-VALUE / ESU R: 0.099 / ESU R Free: 0.092 Details: Hydrogens have been added in their riding positions
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 31.017 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→72.907 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
