 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 7ow9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Crystal structure of a staphylococcal orthologue of CYP134A1 (CYPX) in complex with Cyclo-L-leucyl-L-leucine | |||||||||
 要素 要素 | CYPX | |||||||||
 キーワード キーワード | BIOSYNTHETIC PROTEIN / CYP / Leucine / Cyclic / dipeptide / P450 / cyclodipeptide / pulcherrimin / Pulcherriminic / cypX / CYP134A1 / cyclo-L-leucyl-L-leucyl / Staphylococcus / aureus | |||||||||
| 機能・相同性 | Chem-2IO / PROTOPORPHYRIN IX CONTAINING FE / NITRATE ION 機能・相同性情報 機能・相同性情報 | |||||||||
| 生物種 |   Staphylococcus aureus (黄色ブドウ球菌) Staphylococcus aureus (黄色ブドウ球菌) | |||||||||
| 手法 |  X線回折 / シンクロトロン / 分子置換 / 解像度: 1.8 Å X線回折 / シンクロトロン / 分子置換 / 解像度: 1.8 Å | |||||||||
 データ登録者 データ登録者 | Snee, M. / Levy, C. / Leys, D. / Katariya, M. / Munro, A.W. | |||||||||
| 資金援助 |  英国, 1件 英国, 1件
| |||||||||
 引用 引用 | ジャーナル: To Be Published タイトル: Crystal structure of a staphylococcal orthologue of CYP134A1 (CYPX) in complex with Cyclo-L-leucyl-L-leucine 著者: Snee, M. / Katariya, M. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 7ow9.cif.gz | 262.5 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb7ow9.ent.gz | 214 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 7ow9.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 7ow9_validation.pdf.gz | 376.5 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 7ow9_full_validation.pdf.gz | 378.9 KB | 表示 | |
| XML形式データ | 7ow9_validation.xml.gz | 9.3 KB | 表示 | |
| CIF形式データ | 7ow9_validation.cif.gz | 15.5 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/ow/7ow9ftp://data.pdbj.org/pub/pdb/validation_reports/ow/7ow9 | HTTPS FTP |
-関連構造データ












-リンク
 PDBj
PDBj- 集合体
集合体
| 登録構造単位 | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 45974.688 Da / 分子数: 1 / 由来タイプ: 組換発現 由来: (組換発現) Staphylococcus aureus (黄色ブドウ球菌)株: NCTC 5663 / 発現宿主: |
|---|---|
| #2: 化合物 | ChemComp-2IO / (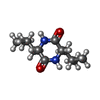  分子量: 226.315 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H22N2O2 / タイプ: SUBJECT OF INVESTIGATION 分子量: 226.315 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C12H22N2O2 / タイプ: SUBJECT OF INVESTIGATION |
| #3: 化合物 | ChemComp-NO3 /   分子量: 62.005 Da / 分子数: 1 / 由来タイプ: 合成 / 式: NO3 分子量: 62.005 Da / 分子数: 1 / 由来タイプ: 合成 / 式: NO3 |
| #4: 化合物 | ChemComp-HEM /   分子量: 616.487 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C34H32FeN4O4 分子量: 616.487 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C34H32FeN4O4 |
| #5: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 293 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 293 / 由来タイプ: 天然 / 式: H2O |
| 研究の焦点であるリガンドがあるか | Y |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.39 Å3/Da / 溶媒含有率: 48.56 % / 解説: Oblong and block-like |
|---|---|
| 結晶化 | 温度: 277.15 K / 手法: 蒸気拡散法, シッティングドロップ法 / pH: 8.5 詳細: 0.2M Ammonium Nitrate, 0.1M Bis Tris Propane pH 8.5, 18% V/V PEG smear High |
-データ収集
| 回折 | 平均測定温度: 100 K / Serial crystal experiment: N |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: Diamond / ビームライン: I04 / 波長: 0.9795 Å |
| 検出器 | タイプ: DECTRIS EIGER2 X 16M / 検出器: PIXEL / 日付: 2020年12月15日 |
| 放射 | プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.9795 Å / 相対比: 1 |
| 反射 | 解像度: 1.8→54.37 Å / Num. obs: 40958 / % possible obs: 99.6 % / 冗長度: 13 % / Biso Wilson estimate: 30.75 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.0751 / Rpim(I) all: 0.021 / Rrim(I) all: 0.078 / Net I/σ(I): 18 |
| 反射 シェル | 解像度: 1.8→1.83 Å / 冗長度: 13.2 % / Rmerge(I) obs: 1.047 / Mean I/σ(I) obs: 0.9 / Num. unique obs: 1997 / CC1/2: 0.914 / Rpim(I) all: 0.295 / % possible all: 97.89 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: 3NC3 解像度: 1.8→52.11 Å / SU ML: 0.2 / 交差検証法: THROUGHOUT / σ(F): 1.37 / 位相誤差: 22.3 / 立体化学のターゲット値: ML
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 減衰半径: 0.9 Å / VDWプローブ半径: 1.11 Å / 溶媒モデル: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso max: 156.37 Å2 / Biso mean: 46.4856 Å2 / Biso min: 22.96 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: final / 解像度: 1.8→52.11 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / Total num. of bins used: 15
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLS | 手法: refined / Origin x: -16.957 Å / Origin y: -22.573 Å / Origin z: -5.749 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 TLSグループ |
|
