Journal: Nat Commun / Year: 2022 Title: Inhibition of SRP-dependent protein secretion by the bacterial alarmone (p)ppGpp. Authors: Laura Czech / Christopher-Nils Mais / Hanna Kratzat / Pinku Sarmah / Pietro Giammarinaro / Sven-Andreas Freibert / Hanna Folke Esser / Joanna Musial / Otto Berninghausen / Wieland Steinchen ...Authors: Laura Czech / Christopher-Nils Mais / Hanna Kratzat / Pinku Sarmah / Pietro Giammarinaro / Sven-Andreas Freibert / Hanna Folke Esser / Joanna Musial / Otto Berninghausen / Wieland Steinchen / Roland Beckmann / Hans-Georg Koch / Gert Bange / Abstract: The stringent response enables bacteria to respond to nutrient limitation and other stress conditions through production of the nucleotide-based second messengers ppGpp and pppGpp, collectively known ...The stringent response enables bacteria to respond to nutrient limitation and other stress conditions through production of the nucleotide-based second messengers ppGpp and pppGpp, collectively known as (p)ppGpp. Here, we report that (p)ppGpp inhibits the signal recognition particle (SRP)-dependent protein targeting pathway, which is essential for membrane protein biogenesis and protein secretion. More specifically, (p)ppGpp binds to the SRP GTPases Ffh and FtsY, and inhibits the formation of the SRP receptor-targeting complex, which is central for the coordinated binding of the translating ribosome to the SecYEG translocon. Cryo-EM analysis of SRP bound to translating ribosomes suggests that (p)ppGpp may induce a distinct conformational stabilization of the NG domain of Ffh and FtsY in Bacillus subtilis but not in E. coli.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 1items
Germany, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



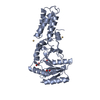













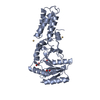
 PDBj
PDBj Assembly
Assembly
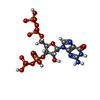
 Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N5O17P4 / Feature type: SUBJECT OF INVESTIGATION
Type: RNA linking / Mass: 603.160 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H17N5O17P4 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 9 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 9 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing