Entry Database : PDB / ID : 7cl9Title Androstenedione-bound structure of CYP154C2 from Streptomyces avermitilis in an open conformation Cytochrome P450 hydroxylase Keywords / / / Function / homology Function Domain/homology Component
/ / / / / / / / Biological species Streptomyces avermitilis MA-4680 = NBRC 14893 (bacteria)Method / / / Resolution : 1.95 Å Authors Xu, L.H. / Fushinobu, S. Funding support Organization Grant number Country National Natural Science Foundation of China (NSFC) 81402810
Journal : To Be Published Title : Androstenedione-bound structure of CYP154C2 from Streptomyces avermitilis in an open conformationAuthors : Xu, L.H. / Fushinobu, S. History Deposition Jul 20, 2020 Deposition site / Processing site Revision 1.0 Jul 21, 2021 Provider / Type Revision 2.0 Sep 20, 2023 Group Atomic model / Author supporting evidence ... Atomic model / Author supporting evidence / Data collection / Database references / Derived calculations / Non-polymer description / Refinement description / Source and taxonomy / Structure summary Category atom_site / chem_comp ... atom_site / chem_comp / chem_comp_atom / chem_comp_bond / database_2 / diffrn_detector / entity / entity_src_gen / pdbx_contact_author / pdbx_entity_instance_feature / pdbx_entity_nonpoly / pdbx_entry_details / pdbx_nonpoly_scheme / pdbx_struct_assembly_gen / pdbx_struct_conn_angle / refine / refine_hist / refine_ls_restr / refine_ls_shell / struct_asym / struct_conn Item _chem_comp.formula / _chem_comp.formula_weight ... _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.mon_nstd_flag / _chem_comp.name / _chem_comp.pdbx_synonyms / _chem_comp.type / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _diffrn_detector.detector / _diffrn_detector.type / _entity_src_gen.pdbx_gene_src_scientific_name / _pdbx_entry_details.has_ligand_of_interest / _pdbx_struct_assembly_gen.asym_id_list / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _refine.B_iso_max / _refine.B_iso_min / _refine.details / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.pdbx_ls_sigma_F / _refine_hist.cycle_id / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_ligand / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_ligand / _refine_hist.pdbx_number_residues_total / _refine_ls_shell.R_factor_R_free_error / _refine_ls_shell.number_reflns_all / _struct_conn.ptnr2_label_asym_id Description / Provider / Type Revision 2.1 Nov 29, 2023 Group / Category
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Streptomyces avermitilis MA-4680 = NBRC 14893 (bacteria)
Streptomyces avermitilis MA-4680 = NBRC 14893 (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors China, 1items
China, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads










 PDBj
PDBj

 Assembly
Assembly

 Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4
Mass: 616.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C34H32FeN4O4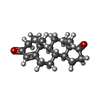
 Mass: 286.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H26O2
Mass: 286.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H26O2
 Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3
Mass: 106.120 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H10O3 Mass: 18.015 Da / Num. of mol.: 304 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 304 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing