 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7bxw | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure ofF RTT109 FROM Candida albicans | |||||||||
 Components Components | Histone acetyltransferase RTT109 | |||||||||
 Keywords Keywords | TRANSFERASE / HISTONE / ACETYLATION / DNA REPLICATION / NUCLEOSOME ASSEMBLY / DNA DAMAGE | |||||||||
| Function / homology |  Function and homology information Function and homology informationphenotypic switching / regulation of phenotypic switching / negative regulation of filamentous growth of a population of unicellular organisms / filamentous growth of a population of unicellular organisms / histone H3K56 acetyltransferase activity / filamentous growth / histone H3 acetyltransferase activity / histone acetyltransferase / DNA damage response / regulation of DNA-templated transcription ...phenotypic switching / regulation of phenotypic switching / negative regulation of filamentous growth of a population of unicellular organisms / filamentous growth of a population of unicellular organisms / histone H3K56 acetyltransferase activity / filamentous growth / histone H3 acetyltransferase activity / histone acetyltransferase / DNA damage response / regulation of DNA-templated transcription / DNA-templated transcription / nucleus Similarity search - Function | |||||||||
| Biological species |  Candida albicans (yeast) Candida albicans (yeast) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77629297561 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.77629297561 Å | |||||||||
 Authors Authors | Lei, J.H. / Chen, Y.P. / Lu, D.R. / Su, D. | |||||||||
| Funding support |  China, 1items China, 1items
| |||||||||
 Citation Citation | Journal: To be published Title: Crystal structure ofF RTT109 FROM Candida albicans Authors: Su, D. / Hu, Q. #1: Journal: J. Biol. Chem / Year: 2011Title: Structure and histone binding properties of the Vps75-Rtt109 chaperone-lysine acetyltransferase complex Authors: Su, D. / Hu, Q. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7bxw.cif.gz | 177.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7bxw.ent.gz | 114.2 KB | Display | PDB format |
| PDBx/mmJSON format | 7bxw.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bx/7bxwftp://data.pdbj.org/pub/pdb/validation_reports/bx/7bxw | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 42028.785 Da / Num. of mol.: 1 / Mutation: S321L,S336L,S339L Source method: isolated from a genetically manipulated source Source: (gene. exp.) Candida albicans (strain SC5314 / ATCC MYA-2876) (yeast)Strain: SC5314 / ATCC MYA-2876 / Gene: RTT109, CAALFM_CR00410WA, CaO19.7491 / Plasmid: pET-Duet1 / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical | 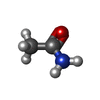  Mass: 59.067 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5NO Mass: 59.067 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H5NO#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 282 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.13 Å3/Da / Density % sol: 42.3 % |
|---|---|
| Crystal grow | Temperature: 289.15 K / Method: vapor diffusion, hanging drop / pH: 6 / Details: 0.1 M Bis-Tris pH 6.0, 15-20% PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 80 K / Ambient temp details: 77 / Serial crystal experiment: N | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRF / Beamline: BL17U1 / Wavelength: 0.975 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: MARMOSAIC 225 mm CCD / Detector: CCD / Date: Apr 5, 2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.975 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.77→50 Å / Num. obs: 33152 / % possible obs: 98.4 % / Redundancy: 7.6 % / Biso Wilson estimate: 22.0078106624 Å2 / Rmerge(I) obs: 0.078 / Rpim(I) all: 0.03 / Rrim(I) all: 0.084 / Χ2: 0.802 / Net I/σ(I): 5.9 / Num. measured all: 252953 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.77629297561→49.5934705553 Å / SU ML: 0.150737498595 / Cross valid method: THROUGHOUT / σ(F): 1.38249054184 / Phase error: 20.9278720425
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.5105658116 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.77629297561→49.5934705553 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
