 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7atg: Crystal structure of Z-DNA in complex with putrescinium and potas... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7atg | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Z-DNA in complex with putrescinium and potassium cations at ultrahigh-resolution | ||||||||||||||||||||||||||||
 Components Components | DNA (5'-D(* Keywords KeywordsDNA / Z-DNA / dual-conformation backbone / putrescinium cation / biogenic polyamines / potassium complex | Function / homology | 4-azaniumylbutylazanium / : / DNA |  Function and homology information Function and homology informationBiological species | synthetic construct (others) | Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 0.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 0.6 Å  Authors AuthorsDrozdzal, P. / Gilski, M. / Jaskolski, M. | Funding support | |  Poland, 1items Poland, 1items
 CitationJournal: Acta Crystallogr.,Sect.B / Year: 2021 CitationJournal: Acta Crystallogr.,Sect.B / Year: 2021Title: Crystal structure of Z-DNA in complex with the polyamine putrescine and potassium cations at ultra-high resolution. Authors: Drozdzal, P. / Gilski, M. / Jaskolski, M. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7atg.cif.gz | 31.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7atg.ent.gz | 21.2 KB | Display | PDB format |
| PDBx/mmJSON format | 7atg.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/at/7atgftp://data.pdbj.org/pub/pdb/validation_reports/at/7atg | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4higS S: Starting model for refinement |
|---|---|
| Similar structure data | |
| Experimental dataset #1 | Data reference: 10.18430/m37atg / Data set type: diffraction image data |







-Links
 PDBj
PDBj

- Assembly
Assembly
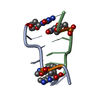
| Deposited unit | 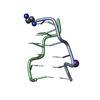
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA chain | Mass: 1810.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #2: Chemical | ChemComp-K / |   Mass: 39.098 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: K / Feature type: SUBJECT OF INVESTIGATION Mass: 39.098 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: K / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-58I / | 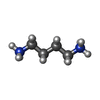  Mass: 90.167 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C4H14N2 / Feature type: SUBJECT OF INVESTIGATION Mass: 90.167 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Formula: C4H14N2 / Feature type: SUBJECT OF INVESTIGATION#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 123 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 123 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.8 Å3/Da / Density % sol: 31.6 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 10%(v/v) (+/-)-2-methyl-2,4-pentanediol (MPD), 40 mM sodium cacodylate pH 6.0, 80 mM KCl, 12 mM NaCl, 14 mM putrescinium dichloride |
-Data collection
| Diffraction |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||
| Detector |
| ||||||||||||||||||
| Radiation |
| ||||||||||||||||||
| Radiation wavelength |
| ||||||||||||||||||
| Reflection | Resolution: 0.6→25.33 Å / Num. obs: 107989 / % possible obs: 90.5 % / Redundancy: 3.7 % / Biso Wilson estimate: 5.17 Å2 / CC1/2: 0.999 / Rmerge(I) obs: 0.024 / Rrim(I) all: 0.027 / Χ2: 1.143 / Net I/σ(I): 29.84 | ||||||||||||||||||
| Reflection shell | Resolution: 0.6→0.61 Å / Redundancy: 1.2 % / Rmerge(I) obs: 0.264 / Mean I/σ(I) obs: 2.88 / Num. unique obs: 1973 / CC1/2: 0.999 / Rrim(I) all: 0.368 / % possible all: 22.6 |

-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4hig Resolution: 0.6→25.33 Å / Num. parameters: 3962 / Num. restraintsaints: 4374 / Cross valid method: FREE R-VALUE Details: THE REFINEMENT WAS CARRIED OUT AGAINST SEPARATE BIJVOET PAIRS. HYDROGEN ATOMS WERE ADDED AT RIDING POSITIONS. THE FINAL REFINEMENT WAS CALCULATED USING WEIGHTED FULL-MATRIX LEAST-SQUARES ...Details: THE REFINEMENT WAS CARRIED OUT AGAINST SEPARATE BIJVOET PAIRS. HYDROGEN ATOMS WERE ADDED AT RIDING POSITIONS. THE FINAL REFINEMENT WAS CALCULATED USING WEIGHTED FULL-MATRIX LEAST-SQUARES PROCEDURE. CONFORMATION-DEPENDENT GEOMETRICAL RESTRAINTS ON BOND LENGTHS AND BOND ANGLES FOR POLYNUCLEOTIDE CHAINS WERE GENERATED USING THE RESTRAINTLIB SERVER: http://achesym.ibch.poznan.pl/restraintlib/
| |||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 36.87 Å2 / Biso mean: 6.26 Å2 / Biso min: 2.73 Å2 | |||||||||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 3 / Occupancy sum hydrogen: 141.29 / Occupancy sum non hydrogen: 333.73 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 0.6→25.33 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
|
