 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7alc | ||||||
|---|---|---|---|---|---|---|---|
| Title | human GCH-GFRP stimulatory complex | ||||||
 Components Components |
| ||||||
 Keywords Keywords | HYDROLASE / GTP cyclohydrolase 1 / GTPCH1 / GTP cyclohydrolase I regulatory protein / GFRP / allosteric regulation / complex / allosteric regulator | ||||||
| Function / homology |  Function and homology information Function and homology informationGTP cyclohydrolase binding / GTP cyclohydrolase I regulator activity / pteridine-containing compound biosynthetic process / negative regulation of biosynthetic process / negative regulation of small molecule metabolic process / regulation of lung blood pressure / GTP cyclohydrolase I / GTP cyclohydrolase I activity / neuromuscular process controlling posture / regulation of removal of superoxide radicals ...GTP cyclohydrolase binding / GTP cyclohydrolase I regulator activity / pteridine-containing compound biosynthetic process / negative regulation of biosynthetic process / negative regulation of small molecule metabolic process / regulation of lung blood pressure / GTP cyclohydrolase I / GTP cyclohydrolase I activity / neuromuscular process controlling posture / regulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / regulation of nitric oxide biosynthetic process / mitogen-activated protein kinase binding / response to pain / dopamine biosynthetic process / neuron projection terminus / response to tumor necrosis factor / tetrahydrofolate biosynthetic process / positive regulation of heart rate / positive regulation of nitric-oxide synthase activity / nitric oxide biosynthetic process / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / response to type II interferon / enzyme inhibitor activity / regulation of blood pressure / melanosome / nuclear membrane / cytoplasmic vesicle / response to lipopolysaccharide / GTPase activity / dendrite / GTP binding / protein homodimerization activity / protein-containing complex / nucleoplasm / zinc ion binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.726 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.726 Å | ||||||
 Authors Authors | Ebenhoch, R. / Nar, H. | ||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2020 Title: A hybrid approach reveals the allosteric regulation of GTP cyclohydrolase I. Authors: Rebecca Ebenhoch / Simone Prinz / Susann Kaltwasser / Deryck J Mills / Robert Meinecke / Martin Rübbelke / Dirk Reinert / Margit Bauer / Lisa Weixler / Markus Zeeb / Janet Vonck / Herbert Nar /  Abstract: Guanosine triphosphate (GTP) cyclohydrolase I (GCH1) catalyzes the conversion of GTP to dihydroneopterin triphosphate (H2NTP), the initiating step in the biosynthesis of tetrahydrobiopterin (BH4). ...Guanosine triphosphate (GTP) cyclohydrolase I (GCH1) catalyzes the conversion of GTP to dihydroneopterin triphosphate (H2NTP), the initiating step in the biosynthesis of tetrahydrobiopterin (BH4). Besides other roles, BH4 functions as cofactor in neurotransmitter biosynthesis. The BH4 biosynthetic pathway and GCH1 have been identified as promising targets to treat pain disorders in patients. The function of mammalian GCH1s is regulated by a metabolic sensing mechanism involving a regulator protein, GCH1 feedback regulatory protein (GFRP). GFRP binds to GCH1 to form inhibited or activated complexes dependent on availability of cofactor ligands, BH4 and phenylalanine, respectively. We determined high-resolution structures of human GCH1-GFRP complexes by cryoelectron microscopy (cryo-EM). Cryo-EM revealed structural flexibility of specific and relevant surface lining loops, which previously was not detected by X-ray crystallography due to crystal packing effects. Further, we studied allosteric regulation of isolated GCH1 by X-ray crystallography. Using the combined structural information, we are able to obtain a comprehensive picture of the mechanism of allosteric regulation. Local rearrangements in the allosteric pocket upon BH4 binding result in drastic changes in the quaternary structure of the enzyme, leading to a more compact, tense form of the inhibited protein, and translocate to the active site, leading to an open, more flexible structure of its surroundings. Inhibition of the enzymatic activity is not a result of hindrance of substrate binding, but rather a consequence of accelerated substrate binding kinetics as shown by saturation transfer difference NMR (STD-NMR) and site-directed mutagenesis. We propose a dissociation rate controlled mechanism of allosteric, noncompetitive inhibition. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7alc.cif.gz | 529.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7alc.ent.gz | 432 KB | Display | PDB format |
| PDBx/mmJSON format | 7alc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/al/7alcftp://data.pdbj.org/pub/pdb/validation_reports/al/7alc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6z80C  6z85C  6z86C  6z87C  6z88C  6z89C  7accC  7al9C  7alaC  7albC  1is8S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 23473.984 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GCH1, DYT5, GCH / Production host:  #2: Protein | Mass: 9992.483 Da / Num. of mol.: 5 / Source method: obtained synthetically / Source: (synth.) Homo sapiens (human) / References: UniProt: P30047#3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION Mass: 65.409 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Zn / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | ChemComp-PHE / 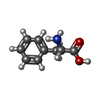  Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C9H11NO2 / Feature type: SUBJECT OF INVESTIGATION Type: L-peptide linking / Mass: 165.189 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C9H11NO2 / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 979 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 979 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.44 Å3/Da / Density % sol: 49.5 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, sitting drop / pH: 6 / Details: 2 M NaCl; 0.1 M NaCit pH 6.0 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X06SA / Wavelength: 1 Å / Beamline: X06SA / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Sep 12, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.726→86.99 Å / Num. obs: 162888 / % possible obs: 99.1 % / Redundancy: 3.5 % / CC1/2: 0.998 / Net I/σ(I): 7.9 |
| Reflection shell | Resolution: 1.726→1.934 Å / Num. unique obs: 44799 / CC1/2: 0.26 / % possible all: 99.7 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1IS8 Resolution: 1.726→86.99 Å / Cor.coef. Fo:Fc: 0.936 / Cor.coef. Fo:Fc free: 0.923 / SU R Cruickshank DPI: 0.172 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.177 / SU Rfree Blow DPI: 0.151 / SU Rfree Cruickshank DPI: 0.15 Details: HYDROGENS WERE FULLY REFINED WITH ZERO OCCUPANCY AT NUCLEAR POSITION. REFINEMENT NOTES. NUMBER OF REFINEMENT NOTES : 1 NOTE 1 : IDEAL-DIST CONTACT TERM CONTACT SETUP. ALL ATOMS HAVE CCP4 ATOM TYPE FROM LIBRARY
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 77.57 Å2 / Biso mean: 28.14 Å2 / Biso min: 5.74 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.28 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.726→86.99 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.73→1.86 Å / Rfactor Rfree error: 0 / Total num. of bins used: 51
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
