GTP cyclohydrolase binding / GTP cyclohydrolase I regulator activity / pteridine-containing compound biosynthetic process / negative regulation of biosynthetic process / negative regulation of small molecule metabolic process / regulation of lung blood pressure / GTP cyclohydrolase I / GTP cyclohydrolase I activity / neuromuscular process controlling posture / regulation of removal of superoxide radicals ...GTP cyclohydrolase binding / GTP cyclohydrolase I regulator activity / pteridine-containing compound biosynthetic process / negative regulation of biosynthetic process / negative regulation of small molecule metabolic process / regulation of lung blood pressure / GTP cyclohydrolase I / GTP cyclohydrolase I activity / neuromuscular process controlling posture / regulation of removal of superoxide radicals / tetrahydrobiopterin biosynthetic process / regulation of nitric oxide biosynthetic process / mitogen-activated protein kinase binding / response to pain / dopamine biosynthetic process / neuron projection terminus / response to tumor necrosis factor / response to type II interferon / tetrahydrofolate biosynthetic process / positive regulation of heart rate / positive regulation of nitric-oxide synthase activity / nitric oxide biosynthetic process / Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation / enzyme inhibitor activity / regulation of blood pressure / melanosome / cytoplasmic vesicle / nuclear membrane / response to lipopolysaccharide / GTPase activity / dendrite / GTP binding / protein homodimerization activity / protein-containing complex / zinc ion binding / nucleoplasm / identical protein binding / nucleus / cytoplasm / cytosol 類似検索 - 分子機能
GTP cyclohydrolase I, feedback regulatory protein / GFRP superfamily / GTP cyclohydrolase I feedback regulatory protein (GFRP) / GTP cyclohydrolase I signature 2. / GTP cyclohydrolase I / GTP cyclohydrolase I, conserved site / GTP cyclohydrolase I domain / GTP cyclohydrolase I, N-terminal domain / GTP cyclohydrolase I / GTP cyclohydrolase I signature 1. / GTP cyclohydrolase I, C-terminal/NADPH-dependent 7-cyano-7-deazaguanine reductase 類似検索 - ドメイン・相同性
ジャーナル: Proc Natl Acad Sci U S A / 年: 2020 タイトル: A hybrid approach reveals the allosteric regulation of GTP cyclohydrolase I. 著者: Rebecca Ebenhoch / Simone Prinz / Susann Kaltwasser / Deryck J Mills / Robert Meinecke / Martin Rübbelke / Dirk Reinert / Margit Bauer / Lisa Weixler / Markus Zeeb / Janet Vonck / Herbert Nar / 要旨: Guanosine triphosphate (GTP) cyclohydrolase I (GCH1) catalyzes the conversion of GTP to dihydroneopterin triphosphate (H2NTP), the initiating step in the biosynthesis of tetrahydrobiopterin (BH4). ...Guanosine triphosphate (GTP) cyclohydrolase I (GCH1) catalyzes the conversion of GTP to dihydroneopterin triphosphate (H2NTP), the initiating step in the biosynthesis of tetrahydrobiopterin (BH4). Besides other roles, BH4 functions as cofactor in neurotransmitter biosynthesis. The BH4 biosynthetic pathway and GCH1 have been identified as promising targets to treat pain disorders in patients. The function of mammalian GCH1s is regulated by a metabolic sensing mechanism involving a regulator protein, GCH1 feedback regulatory protein (GFRP). GFRP binds to GCH1 to form inhibited or activated complexes dependent on availability of cofactor ligands, BH4 and phenylalanine, respectively. We determined high-resolution structures of human GCH1-GFRP complexes by cryoelectron microscopy (cryo-EM). Cryo-EM revealed structural flexibility of specific and relevant surface lining loops, which previously was not detected by X-ray crystallography due to crystal packing effects. Further, we studied allosteric regulation of isolated GCH1 by X-ray crystallography. Using the combined structural information, we are able to obtain a comprehensive picture of the mechanism of allosteric regulation. Local rearrangements in the allosteric pocket upon BH4 binding result in drastic changes in the quaternary structure of the enzyme, leading to a more compact, tense form of the inhibited protein, and translocate to the active site, leading to an open, more flexible structure of its surroundings. Inhibition of the enzymatic activity is not a result of hindrance of substrate binding, but rather a consequence of accelerated substrate binding kinetics as shown by saturation transfer difference NMR (STD-NMR) and site-directed mutagenesis. We propose a dissociation rate controlled mechanism of allosteric, noncompetitive inhibition.
解像度: 1.846→39.74 Å / Cor.coef. Fo:Fc: 0.934 / Cor.coef. Fo:Fc free: 0.913 / SU R Cruickshank DPI: 0.307 / 交差検証法: THROUGHOUT / SU R Blow DPI: 0.316 / SU Rfree Blow DPI: 0.208 / SU Rfree Cruickshank DPI: 0.209
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用
 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード




















 PDBj
PDBj




 集合体
集合体





 分子量: 65.409 Da / 分子数: 5 / 由来タイプ: 合成 / 式: Zn / タイプ: SUBJECT OF INVESTIGATION
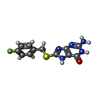
分子量: 65.409 Da / 分子数: 5 / 由来タイプ: 合成 / 式: Zn / タイプ: SUBJECT OF INVESTIGATION 分子量: 291.304 Da / 分子数: 5 / 由来タイプ: 合成 / 式: C12H10FN5OS / タイプ: SUBJECT OF INVESTIGATION
分子量: 291.304 Da / 分子数: 5 / 由来タイプ: 合成 / 式: C12H10FN5OS / タイプ: SUBJECT OF INVESTIGATION 分子量: 22.990 Da / 分子数: 5 / 由来タイプ: 合成 / 式: Na / タイプ: SUBJECT OF INVESTIGATION
分子量: 22.990 Da / 分子数: 5 / 由来タイプ: 合成 / 式: Na / タイプ: SUBJECT OF INVESTIGATION 試料調製
試料調製 / ビームライン: X10SA / 波長: 0.99988 Å
/ ビームライン: X10SA / 波長: 0.99988 Å 解析
解析