 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7ahp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of Ixodes ricinus serpin - Iripin-3 | ||||||
 Components Components | (Putative salivary serpin) x 2 | ||||||
 Keywords Keywords | HYDROLASE INHIBITOR / Serpin / Inhibitor / Ixodes ricinus / HYDROLASE | ||||||
| Function / homology |  Function and homology information Function and homology informationimmune system process / serine-type endopeptidase inhibitor activity / toxin activity / : Similarity search - Function | ||||||
| Biological species |  Ixodes ricinus (castor bean tick) Ixodes ricinus (castor bean tick) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.95 Å | ||||||
 Authors Authors | Kascakova, B. / Kuta Smatanova, I. / Prudnikova, T. | ||||||
| Funding support |  Czech Republic, 1items Czech Republic, 1items
| ||||||
 Citation Citation | Journal: Front Immunol / Year: 2021 Title: Iripin-3, a New Salivary Protein Isolated From Ixodes ricinus Ticks, Displays Immunomodulatory and Anti-Hemostatic Properties In Vitro Authors: Chlastakova, A. / Kotal, J. / Berankova, Z. / Kascakova, B. / Martins, L.A. / Langhansova, H. / Prudnikova, T. / Ederova, M. / Kotsyfakis, M. / Chmelar, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7ahp.cif.gz | 91.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7ahp.ent.gz | Display | PDB format | |
| PDBx/mmJSON format | 7ahp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ah/7ahpftp://data.pdbj.org/pub/pdb/validation_reports/ah/7ahp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3ndaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 39157.332 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Ixodes ricinus (castor bean tick) / Production host:  | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 3712.242 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Ixodes ricinus (castor bean tick) / Production host: | ||||||
| #3: Chemical | 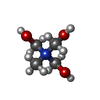  Mass: 122.143 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H12NO3 Mass: 122.143 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H12NO3#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 243 / Source method: isolated from a natural source / Formula: H2OCompound details | Two protein chains result from protease cleavage (between residues 355 and 363) that is common for ...Two protein chains result from protease cleavage (between residues 355 and 363) that is common for serpins. After binding of the serpin reactive center loop (RCL) to protease and subsequent cleavage and insertion of RCL into beta-sheet A. Chain A corresponds to the N-terminal region generated by the proteolytic event and chain lowercase aa corresponds to the C-terminal region. | Has ligand of interest | N | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.6 Å3/Da / Density % sol: 52.68 % |
|---|---|
| Crystal grow | Temperature: 293.15 K / Method: vapor diffusion, sitting drop / pH: 6.5 / Details: potassium thiocyanate, sodium cacodylate, PGA |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jun 17, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→48.32 Å / Num. obs: 34278 / % possible obs: 99.9 % / Redundancy: 1.573 % / CC1/2: 0.998 / Net I/σ(I): 0.1345 |
| Reflection shell | Resolution: 1.95→2.07 Å / Num. unique obs: 889221 / CC1/2: 0.826 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3nda Resolution: 1.95→48.32 Å / Cor.coef. Fo:Fc: 0.945 / Cor.coef. Fo:Fc free: 0.919 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.151 / ESU R Free: 0.138 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 89.72 Å2 / Biso mean: 24.264 Å2 / Biso min: 8.99 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.95→48.32 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.95→1.999 Å / Rfactor Rfree error: 0
|
