 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6zh0: Structure of human galactokinase 1 bound with 2-(4-chlorophenyl)-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6zh0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of human galactokinase 1 bound with 2-(4-chlorophenyl)-N-(pyrimidin-2-yl)acetamide | ||||||
 Components Components | Galactokinase | ||||||
 Keywords Keywords | TRANSFERASE / GALK1 / galactokinase 1 / fragment screening / allosteric fragment / binding hotspt | ||||||
| Function / homology |  Function and homology information Function and homology informationDefective GALK1 causes GALCT2 / galactokinase / galactokinase activity / galactose metabolic process / beta-D-galactose catabolic process via UDP-galactose, Leloir pathway / galactose binding / Galactose catabolism / extracellular exosome / ATP binding / membrane ...Defective GALK1 causes GALCT2 / galactokinase / galactokinase activity / galactose metabolic process / beta-D-galactose catabolic process via UDP-galactose, Leloir pathway / galactose binding / Galactose catabolism / extracellular exosome / ATP binding / membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.5 Å | ||||||
 Authors Authors | Mackinnon, S.R. / Bezerra, G.A. / Zhang, M. / Foster, W. / Krojer, T. / Brandao-Neto, J. / Douangamath, A. / Arrowsmith, C. / Edwards, A. / Bountra, C. ...Mackinnon, S.R. / Bezerra, G.A. / Zhang, M. / Foster, W. / Krojer, T. / Brandao-Neto, J. / Douangamath, A. / Arrowsmith, C. / Edwards, A. / Bountra, C. / Brennan, P. / Lai, K. / Yue, W.W. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2021 Title: Fragment Screening Reveals Starting Points for Rational Design of Galactokinase 1 Inhibitors to Treat Classic Galactosemia. Authors: Mackinnon, S.R. / Krojer, T. / Foster, W.R. / Diaz-Saez, L. / Tang, M. / Huber, K.V.M. / von Delft, F. / Lai, K. / Brennan, P.E. / Arruda Bezerra, G. / Yue, W.W. #1: Journal: Science / Year: 2020Title: In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Authors: Beata Turoňová / Mateusz Sikora / Christoph Schürmann / Wim J H Hagen / Sonja Welsch / Florian E C Blanc / Sören von Bülow / Michael Gecht / Katrin Bagola / Cindy Hörner / Ger van ...Authors: Beata Turoňová / Mateusz Sikora / Christoph Schürmann / Wim J H Hagen / Sonja Welsch / Florian E C Blanc / Sören von Bülow / Michael Gecht / Katrin Bagola / Cindy Hörner / Ger van Zandbergen / Jonathan Landry / Nayara Trevisan Doimo de Azevedo / Shyamal Mosalaganti / Andre Schwarz / Roberto Covino / Michael D Mühlebach / Gerhard Hummer / Jacomine Krijnse Locker / Martin Beck /  Abstract: The spike protein (S) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is required for cell entry and is the primary focus for vaccine development. In this study, we combined cryo- ...The spike protein (S) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is required for cell entry and is the primary focus for vaccine development. In this study, we combined cryo-electron tomography, subtomogram averaging, and molecular dynamics simulations to structurally analyze S in situ. Compared with the recombinant S, the viral S was more heavily glycosylated and occurred mostly in the closed prefusion conformation. We show that the stalk domain of S contains three hinges, giving the head unexpected orientational freedom. We propose that the hinges allow S to scan the host cell surface, shielded from antibodies by an extensive glycan coat. The structure of native S contributes to our understanding of SARS-CoV-2 infection and potentially to the development of safe vaccines. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6zh0.cif.gz | 570.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6zh0.ent.gz | 468.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6zh0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zh/6zh0ftp://data.pdbj.org/pub/pdb/validation_reports/zh/6zh0 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6q3xC  6zfhC  6zgvC  6zgwC  6zgxC  6zgyC  6zgzC  1wuuS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 43102.977 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: GALK1, GALK / Production host:  #2: Sugar | 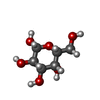  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 3 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 3Source method: isolated from a genetically manipulated source Formula: C6H12O6 #3: Chemical |   Mass: 350.414 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H22N4O2 Mass: 350.414 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C20H22N4O2#4: Chemical | ChemComp-WNP / 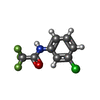  Mass: 223.580 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H5ClF3NO / Feature type: SUBJECT OF INVESTIGATION Mass: 223.580 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H5ClF3NO / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.91 Å3/Da / Density % sol: 57.77 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 0.1 M MOPS/sodium HEPES pH 7.0-7.5, 40-50 % Morpheus Precipitant Mix 4 (50% mix = 12.5% MPD, 12.5% PEG1000, 12.5% PEG3350), 0.1 M Morpheus Carboxylic acids mix (0.02M each of - sodium ...Details: 0.1 M MOPS/sodium HEPES pH 7.0-7.5, 40-50 % Morpheus Precipitant Mix 4 (50% mix = 12.5% MPD, 12.5% PEG1000, 12.5% PEG3350), 0.1 M Morpheus Carboxylic acids mix (0.02M each of - sodium formate, ammonium acetate, sodium citrate tribasic dehydrate, sodium potassium tartrate tetrahydrate and sodium oxamate). PH range: 7.0-7.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I04-1 / Wavelength: 0.91587 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Oct 1, 2018 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.91587 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.27→118.77 Å / Num. obs: 90924 / % possible obs: 99.8 % / Redundancy: 3.5 % / CC1/2: 0.996 / Rmerge(I) obs: 0.13 / Rpim(I) all: 0.082 / Rrim(I) all: 0.154 / Net I/σ(I): 4.7 / Num. measured all: 316693 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1WUU Resolution: 2.5→118.77 Å / Cor.coef. Fo:Fc: 0.929 / Cor.coef. Fo:Fc free: 0.894 / SU B: 33.169 / SU ML: 0.327 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.458 / ESU R Free: 0.307 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : WITH TLS ADDED
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 146.24 Å2 / Biso mean: 50.803 Å2 / Biso min: 32.62 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.5→118.77 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.5→2.565 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
