Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.92 Å / Relative weight: 1
Reflection
Resolution: 1.26→16.66 Å / Num. obs: 35636 / % possible obs: 99.1 % / Redundancy: 6.2 % / CC1/2: 0.999 / Rmerge(I) obs: 0.043 / Net I/σ(I): 27.4
Reflection shell
Resolution: 1.26→1.29 Å / Rmerge(I) obs: 0.551 / Mean I/σ(I) obs: 2.5 / Num. unique obs: 2463 / CC1/2: 0.818 / % possible all: 95.5
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.0257
refinement
XDS
datareduction
Aimless
datascaling
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: in house structure Resolution: 1.26→16.657 Å / Cor.coef. Fo:Fc: 0.966 / Cor.coef. Fo:Fc free: 0.961 / SU B: 0.902 / SU ML: 0.039 / Cross valid method: FREE R-VALUE / ESU R: 0.055 / ESU R Free: 0.055 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Rfree
0.2138
1782
5.01 %
Rwork
0.1957
33784
-
all
0.197
-
-
obs
-
35566
98.858 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL PLUS MASK
Displacement parameters
Biso mean: 19.953 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.482 Å2
0 Å2
0 Å2
2-
-
0.325 Å2
-0 Å2
3-
-
-
-0.807 Å2
Refinement step
Cycle: LAST / Resolution: 1.26→16.657 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1046
0
28
258
1332
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.003
0.013
1132
X-RAY DIFFRACTION
r_bond_other_d
0.001
0.017
1048
X-RAY DIFFRACTION
r_angle_refined_deg
1.159
1.665
1547
X-RAY DIFFRACTION
r_angle_other_deg
1.176
1.614
2468
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
4.919
5
132
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
34.481
25.088
57
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
11.934
15
203
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
9.661
15
3
X-RAY DIFFRACTION
r_chiral_restr
0.055
0.2
146
X-RAY DIFFRACTION
r_gen_planes_refined
0.004
0.02
1214
X-RAY DIFFRACTION
r_gen_planes_other
0.001
0.02
210
X-RAY DIFFRACTION
r_nbd_refined
0.185
0.2
221
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.162
0.2
897
X-RAY DIFFRACTION
r_nbtor_refined
0.169
0.2
532
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.079
0.2
386
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.109
0.2
176
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.182
0.2
12
X-RAY DIFFRACTION
r_nbd_other
0.168
0.2
46
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.064
0.2
36
X-RAY DIFFRACTION
r_mcbond_it
1.132
2.437
507
X-RAY DIFFRACTION
r_mcbond_other
1.132
2.431
506
X-RAY DIFFRACTION
r_mcangle_it
1.923
5.471
634
X-RAY DIFFRACTION
r_mcangle_other
1.921
5.481
635
X-RAY DIFFRACTION
r_scbond_it
1.394
2.648
625
X-RAY DIFFRACTION
r_scbond_other
1.395
2.65
624
X-RAY DIFFRACTION
r_scangle_it
2.353
5.813
909
X-RAY DIFFRACTION
r_scangle_other
2.351
5.812
910
X-RAY DIFFRACTION
r_lrange_it
5.513
24.105
1439
X-RAY DIFFRACTION
r_lrange_other
4.886
22.232
1348
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Rfactor all
Num. reflection all
Fsc free
Fsc work
% reflection obs (%)
WRfactor Rwork
1.26-1.293
0.279
117
0.287
2326
0.286
2575
0.845
0.86
94.8738
0.258
1.293-1.328
0.262
128
0.271
2372
0.271
2561
0.89
0.882
97.6181
0.239
1.328-1.366
0.286
116
0.26
2340
0.262
2481
0.87
0.885
98.9923
0.224
1.366-1.408
0.31
120
0.249
2266
0.252
2395
0.866
0.892
99.6242
0.214
1.408-1.454
0.311
111
0.241
2214
0.244
2336
0.893
0.909
99.5291
0.208
1.454-1.504
0.216
97
0.221
2143
0.221
2249
0.926
0.93
99.5998
0.191
1.504-1.56
0.226
126
0.22
2063
0.22
2201
0.931
0.936
99.4548
0.189
1.56-1.624
0.235
102
0.208
2004
0.209
2112
0.926
0.938
99.7159
0.182
1.624-1.695
0.232
96
0.206
1918
0.207
2023
0.938
0.943
99.5551
0.186
1.695-1.777
0.237
92
0.207
1831
0.208
1931
0.934
0.943
99.5857
0.189
1.777-1.872
0.197
107
0.199
1754
0.199
1868
0.945
0.945
99.6253
0.185
1.872-1.984
0.257
102
0.206
1653
0.209
1759
0.926
0.941
99.7726
0.196
1.984-2.119
0.209
70
0.185
1566
0.186
1654
0.941
0.945
98.9117
0.182
2.119-2.285
0.196
85
0.188
1448
0.188
1551
0.94
0.943
98.8395
0.189
2.285-2.499
0.226
68
0.19
1356
0.192
1428
0.929
0.947
99.7199
0.193
2.499-2.786
0.183
63
0.188
1254
0.188
1321
0.955
0.953
99.6972
0.198
2.786-3.202
0.227
66
0.179
1091
0.181
1164
0.939
0.952
99.3986
0.196
3.202-3.886
0.187
57
0.165
937
0.166
1012
0.964
0.967
98.2213
0.184
3.886-5.352
0.16
37
0.162
751
0.162
814
0.98
0.972
96.8059
0.195
5.352-16.657
0.18
22
0.196
497
0.196
520
0.971
0.966
99.8077
0.229
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj
 Assembly
Assembly

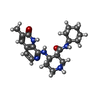
 Mass: 383.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H29N5O2 / Feature type: SUBJECT OF INVESTIGATION
Mass: 383.487 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H29N5O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 18.015 Da / Num. of mol.: 258 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 258 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I04-1 / Wavelength: 0.92 Å
/ Beamline: I04-1 / Wavelength: 0.92 Å Processing
Processing