 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6yma: MicroED structure of acetazolamide-bound human carbonic anhydrase II -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6yma | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | MicroED structure of acetazolamide-bound human carbonic anhydrase II | ||||||||||||
 Components Components | carbonic anhydrase 2 | ||||||||||||
 Keywords Keywords | LYASE / carbonic anhydrase / acetazolamide / inhibitor complex / MicroED | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationpositive regulation of dipeptide transmembrane transport / : / secretion / regulation of monoatomic anion transport / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / positive regulation of synaptic transmission, GABAergic ...positive regulation of dipeptide transmembrane transport / : / secretion / regulation of monoatomic anion transport / cyanamide hydratase / cyanamide hydratase activity / arylesterase activity / regulation of chloride transport / Reversible hydration of carbon dioxide / positive regulation of synaptic transmission, GABAergic / morphogenesis of an epithelium / angiotensin-activated signaling pathway / Developmental Lineage of Pancreatic Ductal Cells / carbonic anhydrase / regulation of intracellular pH / carbonate dehydratase activity / carbon dioxide transport / neuron cellular homeostasis / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / apical part of cell / myelin sheath / extracellular exosome / zinc ion binding / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||
| Method | ELECTRON CRYSTALLOGRAPHY / electron crystallography /  MOLECULAR REPLACEMENT / cryo EM / Resolution: 2.5 Å MOLECULAR REPLACEMENT / cryo EM / Resolution: 2.5 Å | ||||||||||||
 Authors Authors | Clabbers, M.T.B. / Fisher, S.Z. / Coincon, M. / Zou, X. / Xu, H. | ||||||||||||
| Funding support |  Sweden, 3items Sweden, 3items
| ||||||||||||
 Citation Citation | Journal: Commun Biol / Year: 2020 Title: Visualizing drug binding interactions using microcrystal electron diffraction. Authors: Max T B Clabbers / S Zoë Fisher / Mathieu Coinçon / Xiaodong Zou / Hongyi Xu / Abstract: Visualizing ligand binding interactions is important for structure-based drug design and fragment-based screening methods. Rapid and uniform soaking with potentially reduced lattice defects make ...Visualizing ligand binding interactions is important for structure-based drug design and fragment-based screening methods. Rapid and uniform soaking with potentially reduced lattice defects make small macromolecular crystals attractive targets for studying drug binding using microcrystal electron diffraction (MicroED). However, so far no drug binding interactions could unambiguously be resolved by electron diffraction alone. Here, we use MicroED to study the binding of a sulfonamide inhibitor to human carbonic anhydrase isoform II (HCA II). We show that MicroED data can efficiently be collected on a conventional transmission electron microscope from thin hydrated microcrystals soaked with the clinical drug acetazolamide (AZM). The data are of high enough quality to unequivocally fit and resolve the bound inhibitor. We anticipate MicroED can play an important role in facilitating in-house fragment screening for drug discovery, complementing existing methods in structural biology such as X-ray and neutron diffraction. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6yma.cif.gz | 71.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6yma.ent.gz | 41.4 KB | Display | PDB format |
| PDBx/mmJSON format | 6yma.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ym/6ymaftp://data.pdbj.org/pub/pdb/validation_reports/ym/6yma | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6ymbC  3hs4S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data | |
| Experimental dataset #1 | Data reference: 10.15785/SBGRID/792 / Data set type: diffraction image data / Metadata reference: 10.15785/SBGRID/792 |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29289.062 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  |
|---|---|
| #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Zn |
| #3: Chemical | ChemComp-AZM / 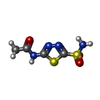  Mass: 222.245 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6N4O3S2 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication*YM Mass: 222.245 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6N4O3S2 / Feature type: SUBJECT OF INVESTIGATION / Comment: medication*YM |
| #4: Chemical | ChemComp-DMS /   Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 27 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 27 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON CRYSTALLOGRAPHY |
|---|---|
| EM experiment | Aggregation state: 3D ARRAY / 3D reconstruction method: electron crystallography |
- Sample preparation
Sample preparation
| Component | Name: Carbonic anhydrase II / Type: COMPLEX / Entity ID: #1 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: Homo sapiens (human) |
| Source (recombinant) | Organism: |
| Buffer solution | pH: 8.5 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Cryogen name: ETHANE |
-Data collection
| Microscopy | Model: JEOL 2100 |
|---|---|
| Electron gun | Electron source: LAB6 / Accelerating voltage: 200 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: DIFFRACTION |
| Image recording | Electron dose: 0.15 e/Å2 / Film or detector model: OTHER |
| EM diffraction | Camera length: 1481.74 mm |
| EM diffraction shell | Resolution: 35.69→2.5 Å / Fourier space coverage: 80 % / Multiplicity: 4.4 / Num. of structure factors: 6895 / Phase residual: 1 ° |
| EM diffraction stats | Fourier space coverage: 80 % / High resolution: 2.5 Å / Num. of intensities measured: 30457 / Num. of structure factors: 6902 / Phase error: 27.44 ° / Phase residual: 1 ° / Phase error rejection criteria: 1 / Rmerge: 27.2 / Rsym: 27.2 |
| Reflection | Biso Wilson estimate: 28.05 Å2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM 3D crystal entity | ∠α: 90 ° / ∠β: 104.62 ° / ∠γ: 90 ° / A: 42.55 Å / B: 41.52 Å / C: 72.11 Å / Space group name: P21 / Space group num: 4 | ||||||||||||||||||||||||||||
| CTF correction | Type: NONE | ||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.5 Å / Resolution method: DIFFRACTION PATTERN/LAYERLINES / Symmetry type: 3D CRYSTAL | ||||||||||||||||||||||||||||
| Atomic model building | Protocol: OTHER / Space: RECIPROCAL | ||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 3HS4 Pdb chain-ID: A / Accession code: 3HS4 / Pdb chain residue range: 4-261 / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3HS4 Resolution: 2.5→35.69 Å / SU ML: 0.3743 / Cross valid method: FREE R-VALUE / σ(F): 1.33 / Phase error: 27.4362 / Stereochemistry target values: CDL v1.2
| ||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | ||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20.65 Å2 | ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell |
|
