 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6yea | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human wtSTING in complex with 2',2'-difluoro-3',3'-cGAMP | ||||||
 Components Components | Stimulator of interferon protein | ||||||
 Keywords Keywords | PROTEIN BINDING / innate immune system / cyclic dinucleotide / STING | ||||||
| Function / homology |  Function and homology information Function and homology information2',3'-cyclic GMP-AMP binding / autophagosome membrane / positive regulation of type I interferon production / endoplasmic reticulum-Golgi intermediate compartment membrane / activation of innate immune response / mitochondrial outer membrane / Golgi membrane / endoplasmic reticulum membrane / perinuclear region of cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.805 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / molecular replacement / Resolution: 2.805 Å | ||||||
 Authors Authors | Boura, E. / Smola, M. | ||||||
 Citation Citation | Journal: Angew.Chem.Int.Ed.Engl. / Year: 2021 Title: Ligand Strain and Its Conformational Complexity Is a Major Factor in the Binding of Cyclic Dinucleotides to STING Protein. Authors: Smola, M. / Gutten, O. / Dejmek, M. / Kozisek, M. / Evangelidis, T. / Tehrani, Z.A. / Novotna, B. / Nencka, R. / Birkus, G. / Rulisek, L. / Boura, E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6yea.cif.gz | 51.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6yea.ent.gz | 34.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6yea.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ye/6yeaftp://data.pdbj.org/pub/pdb/validation_reports/ye/6yea | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6y99C  6ydbC  6ydzC  6z0zC  6z15C  4ksyS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 23189.064 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: STING, LOC340061, hCG_1782396 / Production host:  |
|---|---|
| #2: Chemical | ChemComp-OOE / 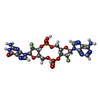  Mass: 678.393 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22F2N10O11P2 / Feature type: SUBJECT OF INVESTIGATION Mass: 678.393 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H22F2N10O11P2 / Feature type: SUBJECT OF INVESTIGATION |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 4 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.36 Å3/Da / Density % sol: 47.91 % |
|---|---|
| Crystal grow | Temperature: 291 K / Method: vapor diffusion, sitting drop Details: 12.5% w/v PEG 1000, 12.5% w/v PEG 3350, 12.5% v/v MPD, 0.2M d-glucose, 0.2M d-mannose, 0.2M d-galactose, 0.2M l-fucose, 0.2M d-xylose, 0.2M N-acetyl-d-glucosamine, 0.1M bicine/Trizma base pH 8.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY  / Beamline: 14.1 / Wavelength: 0.9184 Å / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jan 25, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→35.085 Å / Num. obs: 5820 / % possible obs: 99.74 % / Redundancy: 14 % / CC1/2: 0.989 / Net I/σ(I): 7.1 |
| Reflection shell | Resolution: 2.8→2.906 Å / Mean I/σ(I) obs: 0.86 / Num. unique obs: 554 / CC1/2: 0.465 |
-Phasing
| Phasing | Method: molecular replacement |
|---|
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4KSY Resolution: 2.805→35.085 Å / SU ML: 0.29 / Cross valid method: THROUGHOUT / σ(F): 1.34 / Phase error: 16.9
| ||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||
| Displacement parameters | Biso max: 104 Å2 / Biso mean: 51.4981 Å2 / Biso min: 25.9 Å2 | ||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.805→35.085 Å
| ||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Rfactor Rfree error: 0 / % reflection obs: 100 %
|
