 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6wqh: Molecular basis for the ATPase-powered substrate translocation by... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6wqh | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Molecular basis for the ATPase-powered substrate translocation by the Lon AAA+ protease | ||||||||||||
 Components Components |
| ||||||||||||
 Keywords Keywords | MOTOR PROTEIN / Lon AAA+ / protease / cryo-EM / dual pore-loops | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationendopeptidase La / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / cellular response to heat / sequence-specific DNA binding / serine-type endopeptidase activity / ATP hydrolysis activity / ATP binding / metal ion binding / identical protein binding / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |  Meiothermus taiwanensis (bacteria) Meiothermus taiwanensis (bacteria) | ||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 3.6 Å | ||||||||||||
 Authors Authors | Zhang, K. / Li, S. / Hsiehb, K. / Sub, S. / Pintilie, G. / Chiu, W. / Chang, C. | ||||||||||||
| Funding support |  United States, 3items United States, 3items
| ||||||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2021 Title: Molecular basis for ATPase-powered substrate translocation by the Lon AAA+ protease. Authors: Shanshan Li / Kan-Yen Hsieh / Shih-Chieh Su / Grigore D Pintilie / Kaiming Zhang / Chung-I Chang /   Abstract: The Lon AAA+ (adenosine triphosphatases associated with diverse cellular activities) protease (LonA) converts ATP-fuelled conformational changes into sufficient mechanical force to drive ...The Lon AAA+ (adenosine triphosphatases associated with diverse cellular activities) protease (LonA) converts ATP-fuelled conformational changes into sufficient mechanical force to drive translocation of a substrate into a hexameric proteolytic chamber. To understand the structural basis for the substrate translocation process, we determined the cryo-electron microscopy (cryo-EM) structure of Meiothermus taiwanensis LonA (MtaLonA) in a substrate-engaged state at 3.6 Å resolution. Our data indicate that substrate interactions are mediated by the dual pore loops of the ATPase domains, organized in spiral staircase arrangement from four consecutive protomers in different ATP-binding and hydrolysis states. However, a closed AAA+ ring is maintained by two disengaged ADP-bound protomers transiting between the lowest and highest position. This structure reveals a processive rotary translocation mechanism mediated by LonA-specific nucleotide-dependent allosteric coordination among the ATPase domains, which is induced by substrate binding. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6wqh.cif.gz | 573.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6wqh.ent.gz | 467.7 KB | Display | PDB format |
| PDBx/mmJSON format | 6wqh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/wq/6wqhftp://data.pdbj.org/pub/pdb/validation_reports/wq/6wqh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  21870MC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 88701.766 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Meiothermus taiwanensis (bacteria) / Gene: lonA1, lonProduction host: References: UniProt: A0A059VAZ3, endopeptidase La #2: Protein/peptide | | Mass: 954.168 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: Unfolded substate / Source: (gene. exp.) Production host: #3: Chemical |   Mass: 523.247 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM Mass: 523.247 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H16N5O12P3S / Feature type: SUBJECT OF INVESTIGATION / Comment: ATP-gamma-S, energy-carrying molecule analogue*YM#4: Chemical | ChemComp-4KZ / 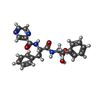  Mass: 418.253 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C22H23BN4O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 418.253 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C22H23BN4O4 / Feature type: SUBJECT OF INVESTIGATION#5: Chemical |   Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Feature type: SUBJECT OF INVESTIGATION / Comment: ADP, energy-carrying molecule*YMHas ligand of interest | Y | Has protein modification | Y | Sequence details | The substrate Ig2 is in the unfolded state, the side chains can not be modeled and are listed as ...The substrate Ig2 is in the unfolded state, the side chains can not be modeled and are listed as UNK in the sequence and coordinates. The complete studied sequence is the following: MTVKPAPSAE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.36 MDa / Experimental value: NO | ||||||||||||||||||||||||
| Source (natural) |
| ||||||||||||||||||||||||
| Source (recombinant) |
| ||||||||||||||||||||||||
| Buffer solution | pH: 8 | ||||||||||||||||||||||||
| Specimen | Conc.: 0.15 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | ||||||||||||||||||||||||
| Specimen support | Grid material: COPPER / Grid mesh size: 200 divisions/in. / Grid type: Quantifoil R1.2/1.3 | ||||||||||||||||||||||||
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 100 % |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Cs: 2.7 mm / C2 aperture diameter: 70 µm |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 6 sec. / Electron dose: 8.5 e/Å2 / Detector mode: COUNTING / Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Num. of real images: 1800 |
| Image scans | Movie frames/image: 30 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C1 (asymmetric) | ||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 3.6 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 23487 / Symmetry type: POINT | ||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 4YPL Accession code: 4YPL / Source name: PDB / Type: experimental model |

