National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI)
United States
Citation
Journal: Nat Commun / Year: 2020 Title: Structural asymmetry governs the assembly and GTPase activity of McrBC restriction complexes. Authors: Yiming Niu / Hiroshi Suzuki / Christopher J Hosford / Thomas Walz / Joshua S Chappie / Abstract: McrBC complexes are motor-driven nucleases functioning in bacterial self-defense by cleaving foreign DNA. The GTP-specific AAA + protein McrB powers translocation along DNA and its hydrolysis ...McrBC complexes are motor-driven nucleases functioning in bacterial self-defense by cleaving foreign DNA. The GTP-specific AAA + protein McrB powers translocation along DNA and its hydrolysis activity is stimulated by its partner nuclease McrC. Here, we report cryo-EM structures of Thermococcus gammatolerans McrB and McrBC, and E. coli McrBC. The McrB hexamers, containing the necessary catalytic machinery for basal GTP hydrolysis, are intrinsically asymmetric. This asymmetry directs McrC binding so that it engages a single active site, where it then uses an arginine/lysine-mediated hydrogen-bonding network to reposition the asparagine in the McrB signature motif for optimal catalytic function. While the two McrBC complexes use different DNA-binding domains, these contribute to the same general GTP-recognition mechanism employed by all G proteins. Asymmetry also induces distinct inter-subunit interactions around the ring, suggesting a coordinated and directional GTP-hydrolysis cycle. Our data provide insights into the conserved molecular mechanisms governing McrB family AAA + motors.
Mass: 539.246 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N5O13P3S / Feature type: SUBJECT OF INVESTIGATION
Has ligand of interest
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.14 Å3/Da / Density % sol: 42.54 %
Crystal grow
Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: 0.1M sodium acetate, pH 6.5, 17.5 % MPD with a drop size of 2 micro-liters and reservoir volume of 650 micro-liters.
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Thermococcus gammatolerans (archaea)
Thermococcus gammatolerans (archaea) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation
 Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj

 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION
Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg / Feature type: SUBJECT OF INVESTIGATION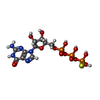
 Mass: 539.246 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N5O13P3S / Feature type: SUBJECT OF INVESTIGATION
Mass: 539.246 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C10H16N5O13P3S / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation Processing
Processing