 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6tp8: Substrate protein interactions in the limbus region of the cataly... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6tp8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Substrate protein interactions in the limbus region of the catalytic site of Candida antarctica Lipase B | |||||||||
 Components Components | Lipase B | |||||||||
 Keywords Keywords | HYDROLASE / Lipase / fatty acid/metabolism / lipid chemistry / interfacial enzymology | |||||||||
| Function / homology |  Function and homology information Function and homology informationtriacylglycerol lipase / triacylglycerol lipase activity / lipid catabolic process Similarity search - Function | |||||||||
| Biological species |  Pseudozyma antarctica (fungus) Pseudozyma antarctica (fungus) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.55 Å | |||||||||
 Authors Authors | Cianci, M. / Silvestrini, L. | |||||||||
 Citation Citation | Journal: Int.J.Biol.Macromol. / Year: 2020 Title: Principles of lipid-enzyme interactions in the limbus region of the catalytic site of Candida antarctica Lipase B. Authors: Silvestrini, L. / Cianci, M. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6tp8.cif.gz | 274.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6tp8.ent.gz | 176 KB | Display | PDB format |
| PDBx/mmJSON format | 6tp8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tp/6tp8ftp://data.pdbj.org/pub/pdb/validation_reports/tp/6tp8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1tcaS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| |||||||||||||||
| 2 | 
| |||||||||||||||
| Unit cell |
| |||||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 33040.238 Da / Num. of mol.: 3 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Pseudozyma antarctica (fungus) / Production host: #2: Polysaccharide | Source method: isolated from a genetically manipulated source #3: Chemical |   Mass: 138.102 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H11O3P / Feature type: SUBJECT OF INVESTIGATION Mass: 138.102 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C4H11O3P / Feature type: SUBJECT OF INVESTIGATION#4: Chemical | 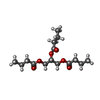  Mass: 302.363 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H26O6 / Feature type: SUBJECT OF INVESTIGATION Mass: 302.363 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H26O6 / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 1461 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 1461 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.45 Å3/Da / Density % sol: 49.7 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 4.8 Details: Crystallization trials were performed at 293 K using the hanging-drop method using a Qiagen EasyXtal 15-well plate. A 15 mg.mL CALB solution in 20mM Na(CH3COO) pH = 4.8 was incubated with a ...Details: Crystallization trials were performed at 293 K using the hanging-drop method using a Qiagen EasyXtal 15-well plate. A 15 mg.mL CALB solution in 20mM Na(CH3COO) pH = 4.8 was incubated with a solution of paraoxon-ethyl (SIGMA catalog n. N12816) 30mM in water for one day. 1 uL of CALB solution was diluted with 1 uL of the precipitant solution, made of 200mM Na(CH3COO) pH = 4.8, 20% (w.v) PEG4000, and 15% (v.v) glyceryl tributyrate (SIGMA catalog n. T8626). The drop was equilibrated by vapor diffusion against 500 uL of the precipitant solution. Protein crystals of the CALB complex appeared within two weeks and grew to a size of 0.1 x 0.1 x 0.05 mm3 in five weeks. |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X13 / Wavelength: 0.824 Å / Beamline: X13 / Wavelength: 0.824 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 22, 2013 |
| Radiation | Monochromator: Si (III) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.824 Å / Relative weight: 1 |
| Reflection | Resolution: 1.55→67.69 Å / Num. obs: 138806 / % possible obs: 99.18 % / Observed criterion σ(I): 1 / Redundancy: 7.6 % / Biso Wilson estimate: 18.9 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.121 / Rpim(I) all: 0.05 / Rrim(I) all: 0.14 / Net I/σ(I): 8.4 |
| Reflection shell | Resolution: 1.55→1.605 Å / Redundancy: 7.2 % / Rmerge(I) obs: 1.46 / Mean I/σ(I) obs: 1 / Num. unique obs: 48047 / CC1/2: 0.55 / Rpim(I) all: 0.877 / Rrim(I) all: 1.7 / % possible all: 98.05 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1TCA Resolution: 1.55→67.69 Å / SU ML: 0.19 / Cross valid method: FREE R-VALUE / σ(F): 1.34 / Phase error: 21.037
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 21.85 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.55→67.69 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
