Entry Database : PDB / ID : 6tneTitle Crystal structure of receiver domain from Hybrid Histidine Kinase CckA Histidine kinase Keywords / / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / Biological species Caulobacter vibrioides (bacteria)Method / / / / Resolution : 1.25 Å Authors Dubey, B.N. / Bruederlin, M. / Schirmer, T. Funding support Organization Grant number Country Swiss National Science Foundation 31003A-166652
Journal : Nat Commun / Year : 2023Title : Structural features discriminating hybrid histidine kinase Rec domains from response regulator homologs.Authors : Bruderlin, M. / Bohm, R. / Fadel, F. / Hiller, S. / Schirmer, T. / Dubey, B.N. History Deposition Dec 6, 2019 Deposition site / Processing site Revision 1.0 Dec 16, 2020 Provider / Type Revision 2.0 Oct 5, 2022 Group Advisory / Atomic model ... Advisory / Atomic model / Data collection / Database references / Derived calculations / Non-polymer description / Other / Polymer sequence / Refinement description / Source and taxonomy / Structure summary Category atom_site / atom_site_anisotrop ... atom_site / atom_site_anisotrop / atom_sites / chem_comp / database_2 / entity / entity_poly / entity_poly_seq / entity_src_gen / pdbx_contact_author / pdbx_distant_solvent_atoms / pdbx_nonpoly_scheme / pdbx_poly_seq_scheme / pdbx_refine_tls / pdbx_refine_tls_group / pdbx_struct_sheet_hbond / pdbx_unobs_or_zero_occ_residues / pdbx_validate_close_contact / pdbx_validate_rmsd_angle / pdbx_validate_rmsd_bond / pdbx_validate_symm_contact / refine / refine_hist / refine_ls_restr / refine_ls_shell / reflns / reflns_shell / software / struct_conf / struct_mon_prot_cis / struct_ref / struct_ref_seq / struct_ref_seq_dif / struct_sheet_range Item _atom_sites.fract_transf_matrix[1][3] / _chem_comp.formula ... _atom_sites.fract_transf_matrix[1][3] / _chem_comp.formula / _chem_comp.formula_weight / _chem_comp.id / _chem_comp.mon_nstd_flag / _chem_comp.name / _chem_comp.type / _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _entity.formula_weight / _entity.pdbx_description / _entity.pdbx_ec / _entity.pdbx_number_of_molecules / _entity_poly.pdbx_seq_one_letter_code / _entity_poly.pdbx_seq_one_letter_code_can / _entity_src_gen.pdbx_end_seq_num / _pdbx_contact_author.id / _pdbx_struct_sheet_hbond.range_1_auth_comp_id / _pdbx_struct_sheet_hbond.range_1_auth_seq_id / _pdbx_struct_sheet_hbond.range_1_label_comp_id / _pdbx_struct_sheet_hbond.range_1_label_seq_id / _pdbx_struct_sheet_hbond.range_2_label_seq_id / _pdbx_validate_symm_contact.auth_atom_id_1 / _pdbx_validate_symm_contact.auth_comp_id_1 / _pdbx_validate_symm_contact.auth_seq_id_1 / _pdbx_validate_symm_contact.auth_seq_id_2 / _pdbx_validate_symm_contact.dist / _pdbx_validate_symm_contact.site_symmetry_2 / _refine.B_iso_max / _refine.B_iso_mean / _refine.B_iso_min / _refine.aniso_B[1][1] / _refine.aniso_B[1][2] / _refine.aniso_B[1][3] / _refine.aniso_B[2][2] / _refine.aniso_B[2][3] / _refine.aniso_B[3][3] / _refine.correlation_coeff_Fo_to_Fc / _refine.correlation_coeff_Fo_to_Fc_free / _refine.details / _refine.ls_R_factor_R_free / _refine.ls_R_factor_R_work / _refine.ls_R_factor_obs / _refine.ls_d_res_low / _refine.ls_number_reflns_R_free / _refine.ls_number_reflns_R_work / _refine.ls_number_reflns_obs / _refine.ls_percent_reflns_R_free / _refine.overall_SU_B / _refine.overall_SU_ML / _refine.overall_SU_R_Cruickshank_DPI / _refine.pdbx_ls_sigma_F / _refine.pdbx_overall_ESU_R / _refine.pdbx_overall_ESU_R_Free / _refine.pdbx_overall_phase_error / _refine.pdbx_solvent_ion_probe_radii / _refine.pdbx_solvent_shrinkage_radii / _refine.pdbx_solvent_vdw_probe_radii / _refine.pdbx_stereochemistry_target_values / _refine.solvent_model_details / _refine_hist.d_res_low / _refine_hist.number_atoms_solvent / _refine_hist.number_atoms_total / _refine_hist.pdbx_B_iso_mean_solvent / _refine_hist.pdbx_number_atoms_protein / _refine_hist.pdbx_number_residues_total / _reflns.B_iso_Wilson_estimate / _reflns.d_resolution_low / _reflns.number_obs / _reflns.pdbx_Rmerge_I_obs / _reflns.pdbx_Rpim_I_all / _reflns.pdbx_Rrim_I_all / _reflns.pdbx_netI_over_sigmaI / _reflns.pdbx_number_measured_all / _reflns.pdbx_redundancy / _reflns.pdbx_scaling_rejects / _reflns.percent_possible_obs / _software.classification / _software.contact_author / _software.contact_author_email / _software.language / _software.location / _software.name / _software.type / _software.version / _struct_conf.beg_label_seq_id / _struct_conf.end_auth_comp_id / _struct_conf.end_auth_seq_id / _struct_conf.end_label_comp_id / _struct_conf.end_label_seq_id / _struct_conf.pdbx_PDB_helix_length / _struct_mon_prot_cis.label_seq_id / _struct_mon_prot_cis.pdbx_label_seq_id_2 / _struct_mon_prot_cis.pdbx_omega_angle / _struct_ref.pdbx_align_begin / _struct_ref.pdbx_seq_one_letter_code / _struct_ref_seq.db_align_beg / _struct_ref_seq.db_align_end / _struct_ref_seq.pdbx_auth_seq_align_beg / _struct_ref_seq.pdbx_auth_seq_align_end / _struct_ref_seq.seq_align_beg / _struct_ref_seq.seq_align_end / _struct_sheet_range.beg_label_seq_id / _struct_sheet_range.end_label_seq_id Description / Provider / Type Revision 2.1 May 1, 2024 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_modelRevision 2.2 Sep 10, 2025 Group / Structure summary / Category / citation_author / pdbx_entry_detailsItem _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.journal_volume / _citation.page_first / _citation.page_last / _citation.pdbx_database_id_DOI / _citation.pdbx_database_id_PubMed / _citation.title / _citation.year / _citation_author.identifier_ORCID / _citation_author.name
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Caulobacter vibrioides (bacteria)
Caulobacter vibrioides (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Switzerland, 1items
Switzerland, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
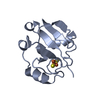








 PDBj
PDBj


 Assembly
Assembly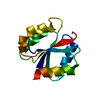
 Mass: 18.015 Da / Num. of mol.: 103 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 103 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing