 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6tlk: Structure of methylene-tetrahydromethanopterin dehydrogenase from... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6tlk | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of methylene-tetrahydromethanopterin dehydrogenase from Methylorubrum extorquens AM1 in an open conformation containing NADP+ and methylene-H4MPT | ||||||||||||
 Components Components | Bifunctional protein MdtA | ||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / One-carbon metabolism / Enzyme catalysis / Dehydrogenase / Conformational changes / Methylotrophy | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationOxidoreductases; Acting on the CH-NH group of donors; With NAD+ or NADP+ as acceptor / methylenetetrahydrofolate dehydrogenase (NADP+) / methylenetetrahydrofolate dehydrogenase (NADP+) activity / formaldehyde catabolic process / one-carbon metabolic process / cytoplasm Similarity search - Function | ||||||||||||
| Biological species |  Methylorubrum extorquens AM1 (bacteria) Methylorubrum extorquens AM1 (bacteria) | ||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||||||||
 Authors Authors | Wagner, T. / Huang, G. / Demmer, U. / Warkentin, E. / Ermler, U. / Shima, S. | ||||||||||||
| Funding support |  Germany, Germany,  China, 3items China, 3items
| ||||||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2020 Title: The Hydride Transfer Process in NADP-dependent Methylene-tetrahydromethanopterin Dehydrogenase. Authors: Huang, G. / Wagner, T. / Demmer, U. / Warkentin, E. / Ermler, U. / Shima, S. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6tlk.cif.gz | 691.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6tlk.ent.gz | 570.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6tlk.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/tl/6tlkftp://data.pdbj.org/pub/pdb/validation_reports/tl/6tlk | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6tgeC  6tm3C  6yk9C  6ykaC  6ykbC  1lu9S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 6 molecules ABCDEF
| #1: Protein | Mass: 31512.734 Da / Num. of mol.: 6 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Methylorubrum extorquens AM1 (bacteria)Gene: mtdA, MexAM1_META1p1728 / Plasmid: pET-24b(+) Details (production host): The synthesized DNA fragment was inserted into the expression vector pET-24b (+) at the NdeI and SalI restriction-enzyme digestion-sites Production host: References: UniProt: P55818, Oxidoreductases; Acting on the CH-NH group of donors; With NAD+ or NADP+ as acceptor, methylenetetrahydrofolate dehydrogenase (NADP+) |
|---|
-Non-polymers , 7 types, 2122 molecules 

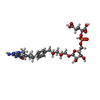




| #2: Chemical | ChemComp-NAP /  Mass: 743.405 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C21H28N7O17P3 / Feature type: SUBJECT OF INVESTIGATION Mass: 743.405 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C21H28N7O17P3 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical | ChemComp-FMT /  Mass: 46.025 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: CH2O2 Mass: 46.025 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: CH2O2#4: Chemical |  Mass: 788.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C31H45N6O16P / Feature type: SUBJECT OF INVESTIGATION Mass: 788.693 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C31H45N6O16P / Feature type: SUBJECT OF INVESTIGATION#5: Chemical | ChemComp-GOL /  Mass: 92.094 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 6 / Source method: isolated from a natural source / Formula: C3H8O3#6: Chemical | ChemComp-CL / |  Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl#7: Chemical | ChemComp-MG / |  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 2093 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.52 Å3/Da / Density % sol: 51.14 % / Description: Thick plate. |
|---|---|
| Crystal grow | Temperature: 281.15 K / Method: vapor diffusion, sitting drop / pH: 7.5 Details: The protein at 25 mg/ml was in 25 mM tris(hydroxymethyl)aminomethane (Tris)/HCl buffer pH 7.5 containing 150 mM NaCl, 5% glycerol and 2 mM dithiothreitol, supplemented with 2.5 mM methenyl- ...Details: The protein at 25 mg/ml was in 25 mM tris(hydroxymethyl)aminomethane (Tris)/HCl buffer pH 7.5 containing 150 mM NaCl, 5% glycerol and 2 mM dithiothreitol, supplemented with 2.5 mM methenyl-H4MPT and 2 mM NADP. The crystallization solution was 20% w/v polyethylene glycol 3350 and 200 mM magnesium formate. The crystallization drop contained 0.7 ul of protein solution and 0.7 ul crystallization solution PH range: / / Temp details: / |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM30A / Wavelength: 0.97992 Å / Beamline: BM30A / Wavelength: 0.97992 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Sep 29, 2017 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 0.97992 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 1.8→78.401 Å / Num. obs: 161189 / % possible obs: 98.9 % / Redundancy: 3 % / Biso Wilson estimate: 28.48 Å2 / CC1/2: 0.99 / Rmerge(I) obs: 0.066 / Rpim(I) all: 0.044 / Rrim(I) all: 0.08 / Rsym value: 0.066 / Net I/av σ(I): 10.5 / Net I/σ(I): 12.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 1LU9 Resolution: 1.8→26.35 Å / Cor.coef. Fo:Fc: 0.962 / Cor.coef. Fo:Fc free: 0.949 / SU R Cruickshank DPI: 0.167 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.126 / SU Rfree Blow DPI: 0.116 / SU Rfree Cruickshank DPI: 0.111
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 136.77 Å2 / Biso mean: 36.17 Å2 / Biso min: 12.8 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.22 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 1.8→26.35 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.8→1.85 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
