 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6srj: X-ray pump X-ray probe on thaumatin nanocrystals: single pulse re... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6srj | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray pump X-ray probe on thaumatin nanocrystals: single pulse reference data | ||||||
 Components Components | Thaumatin-1 | ||||||
 Keywords Keywords | PLANT PROTEIN / Radiation damage / SERIAL femtosecond CRYSTALLOGRAPHY / X-ray FREE-ELECTRON LASER / time-resolved crystallography | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  Thaumatococcus daniellii (katemfe) Thaumatococcus daniellii (katemfe) | ||||||
| Method |  X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / FREE ELECTRON LASER / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Kloos, M. / Gorel, A. / Nass, K. | ||||||
 Citation Citation | Journal: Nat Commun / Year: 2020 Title: Structural dynamics in proteins induced by and probed with X-ray free-electron laser pulses. Authors: Nass, K. / Gorel, A. / Abdullah, M.M. / V Martin, A. / Kloos, M. / Marinelli, A. / Aquila, A. / Barends, T.R.M. / Decker, F.J. / Bruce Doak, R. / Foucar, L. / Hartmann, E. / Hilpert, M. / ...Authors: Nass, K. / Gorel, A. / Abdullah, M.M. / V Martin, A. / Kloos, M. / Marinelli, A. / Aquila, A. / Barends, T.R.M. / Decker, F.J. / Bruce Doak, R. / Foucar, L. / Hartmann, E. / Hilpert, M. / Hunter, M.S. / Jurek, Z. / Koglin, J.E. / Kozlov, A. / Lutman, A.A. / Kovacs, G.N. / Roome, C.M. / Shoeman, R.L. / Santra, R. / Quiney, H.M. / Ziaja, B. / Boutet, S. / Schlichting, I. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6srj.cif.gz | 56.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6srj.ent.gz | 39.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6srj.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 6srj_validation.pdf.gz | 440.5 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 6srj_full_validation.pdf.gz | 443.1 KB | Display | |
| Data in XML | 6srj_validation.xml.gz | 11 KB | Display | |
| Data in CIF | 6srj_validation.cif.gz | 14.9 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sr/6srjftp://data.pdbj.org/pub/pdb/validation_reports/sr/6srj | HTTPS FTP |
-Related structure data
| Related structure data |  6sr0C  6sr1C  6sr2C  6sr3C  6sr4C  6sr5C  6srkC  6srlC  6sroC  6srpC  6srqC  5fgtS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22227.059 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Thaumatococcus daniellii (katemfe) / References: UniProt: P02883 |
|---|---|
| #2: Chemical | ChemComp-TLA / 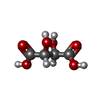  Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O6 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 97 / Source method: isolated from a natural source / Formula: H2O |
| Has ligand of interest | N |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.47 Å3/Da / Density % sol: 50.19 % / Description: nanocrystals |
|---|---|
| Crystal grow | Temperature: 293 K / Method: batch mode / pH: 7 / Details: 0.8 M NA, K TARTRATE, 0.1 M NA HEPES PH 7.0 |
-Data collection
| Diffraction | Mean temperature: 293 K / Serial crystal experiment: Y |
|---|---|
| Diffraction source | Source: FREE ELECTRON LASER / Site: SLAC LCLS  / Beamline: CXI / Wavelength: 1.75 Å / Beamline: CXI / Wavelength: 1.75 Å |
| Detector | Type: CS-PAD CXI-1 / Detector: PIXEL / Date: Feb 15, 2015 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.75 Å / Relative weight: 1 |
| Reflection | Resolution: 2.32→23 Å / Num. obs: 11104 / % possible obs: 100 % / Redundancy: 1 % / CC1/2: 0.973 / R split: 0.09 / Net I/σ(I): 7.4 |
| Reflection shell | Resolution: 2.32→2.36 Å / Mean I/σ(I) obs: 5.2 / Num. unique obs: 850 / CC1/2: 0.973 / R split: 0.222 |
| Serial crystallography measurement | Focal spot size: 0.025 µm2 / Pulse duration: 15 fsec. / Pulse energy: 500 µJ / Pulse photon energy: 7.07 keV / XFEL pulse repetition rate: 120 Hz |
| Serial crystallography sample delivery | Description: GDVN injection / Method: injection |
| Serial crystallography data reduction | Frames indexed: 11000 / Lattices indexed: 11000 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5fgt Resolution: 2.3→23 Å / Cor.coef. Fo:Fc: 0.971 / Cor.coef. Fo:Fc free: 0.953 / SU B: 3.829 / SU ML: 0.092 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.201 / ESU R Free: 0.156 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY. The submitted coordinates represent the mean of 100 coordinate sets obtained by refining against 100 ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY. The submitted coordinates represent the mean of 100 coordinate sets obtained by refining against 100 different integrated diffraction intensity datasets obtained by jackknife resampling of the diffraction snapshots. The coordinate header originates from one of the individual refinements. Cysteine residues were modeled as alanines and non-covalently attached sulphur atoms.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 122.18 Å2 / Biso mean: 26.368 Å2 / Biso min: 13.64 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.3→23 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.36 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
|
