ALIGNMENT: - Global alignment of extracted AA sequence from the pdb file with the original sequence ...ALIGNMENT: - Global alignment of extracted AA sequence from the pdb file with the original sequence inserted in the template - Be aware that the alignment is the best one for the given scoring schemata but does not have to be the best one out of all kinds of possible alignments! - No substitution matrix used. Just matches, mismatches and gaps ar considered - The first sequence was extracted from the xml-template file according to the chosen template. The second sequence was extract from the ATOM section of the pdb file. GNPLVYLDVDANGKPLGRVVLELKADVVPKTAENFRALCTGEKGFGYKGSTFHRVIPSFMCQAGD GNPLVYLDVDANGKPLGR-VLELKAD-VPKTAENFRALCTGEKGFGYKGSTFHRVIPSFMCQAGD FTNHNGTGGKSIYGSRFPDENFTLKHVGPGVLSMANAGPNTNGSQFFICTIKTDWLDGKHVVFGH FTNHNGT-GKSIYGSRFPDENFTLKHVGPGVLSMANAGPNTNGSQ-FICTIKTDWLDGKH-VFGH VIEGMDVVKKIESFGSKSGRTSKKIVITDCGQLS VIEGMD-V-KIESFGSKSGRTS-KIVITDCGQLS
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Homo sapiens (ヒト)
Homo sapiens (ヒト) X線回折 /
X線回折 /  データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード
















 PDBj
PDBj

 集合体
集合体

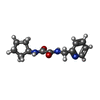
 分子量: 247.293 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C13H17N3O2
分子量: 247.293 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C13H17N3O2 分子量: 18.015 Da / 分子数: 333 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 333 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 解析
解析