 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6pek: Structure of Spastin Hexamer (Subunit A-E) in complex with substr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6pek | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of Spastin Hexamer (Subunit A-E) in complex with substrate peptide | |||||||||||||||||||||||||||||||||||||||||||||||||||
 Components Components |
| |||||||||||||||||||||||||||||||||||||||||||||||||||
 Keywords Keywords | MOTOR PROTEIN / AAA+ ATPase / Microtubule Severing | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationcytokinetic process / microtubule-severing ATPase / microtubule severing ATPase activity / central nervous system neuron axonogenesis / microtubule severing / positive regulation of microtubule depolymerization / endoplasmic reticulum tubular network / exit from mitosis / cytoskeleton-dependent cytokinesis / nuclear membrane reassembly ...cytokinetic process / microtubule-severing ATPase / microtubule severing ATPase activity / central nervous system neuron axonogenesis / microtubule severing / positive regulation of microtubule depolymerization / endoplasmic reticulum tubular network / exit from mitosis / cytoskeleton-dependent cytokinesis / nuclear membrane reassembly / mitotic nuclear membrane reassembly / Sealing of the nuclear envelope (NE) by ESCRT-III / membrane fission / anterograde axonal transport / microtubule bundle formation / protein hexamerization / mitotic spindle disassembly / axonal transport of mitochondrion / positive regulation of cytokinesis / mitotic cytokinesis / endoplasmic reticulum to Golgi vesicle-mediated transport / alpha-tubulin binding / beta-tubulin binding / axon cytoplasm / lipid droplet / protein homooligomerization / spindle / spindle pole / nuclear membrane / cytoplasmic vesicle / midbody / microtubule binding / microtubule / endosome / axon / centrosome / endoplasmic reticulum membrane / protein-containing complex binding / perinuclear region of cytoplasm / ATP hydrolysis activity / extracellular exosome / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human)synthetic construct (others) | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 4.2 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||
 Authors Authors | Han, H. / Schubert, H.L. / McCullough, J. / Monroe, N. / Sundquist, W.I. / Hill, C.P. | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Funding support |  United States, 1items United States, 1items
| |||||||||||||||||||||||||||||||||||||||||||||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2020 Title: Structure of spastin bound to a glutamate-rich peptide implies a hand-over-hand mechanism of substrate translocation. Authors: Han Han / Heidi L Schubert / John McCullough / Nicole Monroe / Michael D Purdy / Mark Yeager / Wesley I Sundquist / Christopher P Hill / Abstract: Many members of the AAA+ ATPase family function as hexamers that unfold their protein substrates. These AAA unfoldases include spastin, which plays a critical role in the architecture of eukaryotic ...Many members of the AAA+ ATPase family function as hexamers that unfold their protein substrates. These AAA unfoldases include spastin, which plays a critical role in the architecture of eukaryotic cells by driving the remodeling and severing of microtubules, which are cytoskeletal polymers of tubulin subunits. Here, we demonstrate that a human spastin binds weakly to unmodified peptides from the C-terminal segment of human tubulin α1A/B. A peptide comprising alternating glutamate and tyrosine residues binds more tightly, which is consistent with the known importance of glutamylation for spastin microtubule severing activity. A cryo-EM structure of the spastin-peptide complex at 4.2 Å resolution revealed an asymmetric hexamer in which five spastin subunits adopt a helical, spiral staircase configuration that binds the peptide within the central pore, whereas the sixth subunit of the hexamer is displaced from the peptide/substrate, as if transitioning from one end of the helix to the other. This configuration differs from a recently published structure of spastin from , which forms a six-subunit spiral without a transitioning subunit. Our structure resembles other recently reported AAA unfoldases, including the meiotic clade relative Vps4, and supports a model in which spastin utilizes a hand-over-hand mechanism of tubulin translocation and microtubule remodeling. | |||||||||||||||||||||||||||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6pek.cif.gz | 264.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6pek.ent.gz | 206 KB | Display | PDB format |
| PDBx/mmJSON format | 6pek.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pe/6pekftp://data.pdbj.org/pub/pdb/validation_reports/pe/6pek | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  20327MC  6penC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10382 (Title: Structure of spastin bound to a glutamate-rich peptide implies a hand-over-hand mechanism of substrate translocation. Data size: 1.8 TB Data #1: Unaligned multi-frame movies of ADPBeFx-bound Spastin [micrographs - multiframe]) |








-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 54498.328 Da / Num. of mol.: 5 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: SPAST, ADPSP, FSP2, KIAA1083, SPG4 / Production host:  #2: Protein/peptide | | Mass: 1479.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #3: Chemical | ChemComp-ADP /   Mass: 427.201 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#4: Chemical | ChemComp-BEF / 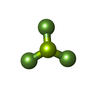  Mass: 66.007 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: BeF3 Mass: 66.007 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: BeF3#5: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: MgHas ligand of interest | N | Has protein modification | N | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Spastin Hexamer in complex with substrate peptide / Type: COMPLEX / Entity ID: #1-#2 / Source: MULTIPLE SOURCES |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: Homo sapiens (human) |
| Buffer solution | pH: 8 |
| Specimen | Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: FEI TITAN KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD |
| Image recording | Electron dose: 57 e/Å2 / Film or detector model: FEI FALCON III (4k x 4k) |
- Processing
Processing
| Software | Name: PHENIX / Version: 1.16_3549: / Classification: refinement | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software | Name: PHENIX / Category: model refinement | ||||||||||||||||||||||||
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 4.2 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 119984 / Symmetry type: POINT | ||||||||||||||||||||||||
| Refine LS restraints |
|
