 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
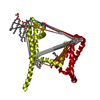
Yorodumi- PDB-6p6y: Crystal structure of voltage-gated sodium channel NavAb V100C/Q15... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6p6y | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal structure of voltage-gated sodium channel NavAb V100C/Q150C disulfide crosslinked mutant in the activated state | |||||||||||||||
 Components Components | Ion transport protein | |||||||||||||||
 Keywords Keywords | membrane protein / metal transport / Ion channel / ion transport protein | |||||||||||||||
| Function / homology |  Function and homology information Function and homology informationvoltage-gated sodium channel complex / voltage-gated sodium channel activity / metal ion binding / identical protein binding Similarity search - Function | |||||||||||||||
| Biological species |  Arcobacter butzleri (bacteria) Arcobacter butzleri (bacteria) | |||||||||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.89 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.89 Å | |||||||||||||||
 Authors Authors | Wisedchaisri, G. / Tonggu, L. / McCord, E. / Gamal El-din, T.M. / Wang, L. / Zheng, N. / Catterall, W.A. | |||||||||||||||
| Funding support |  United States, 4items United States, 4items
| |||||||||||||||
 Citation Citation | Journal: Cell / Year: 2019 Title: Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Authors: Goragot Wisedchaisri / Lige Tonggu / Eedann McCord / Tamer M Gamal El-Din / Liguo Wang / Ning Zheng / William A Catterall / Abstract: Voltage-gated sodium (Na) channels initiate action potentials in nerve, muscle, and other electrically excitable cells. The structural basis of voltage gating is uncertain because the resting state ...Voltage-gated sodium (Na) channels initiate action potentials in nerve, muscle, and other electrically excitable cells. The structural basis of voltage gating is uncertain because the resting state exists only at deeply negative membrane potentials. To stabilize the resting conformation, we inserted voltage-shifting mutations and introduced a disulfide crosslink in the VS of the ancestral bacterial sodium channel NaAb. Here, we present a cryo-EM structure of the resting state and a complete voltage-dependent gating mechanism. The S4 segment of the VS is drawn intracellularly, with three gating charges passing through the transmembrane electric field. This movement forms an elbow connecting S4 to the S4-S5 linker, tightens the collar around the S6 activation gate, and prevents its opening. Our structure supports the classical "sliding helix" mechanism of voltage sensing and provides a complete gating mechanism for voltage sensor function, pore opening, and activation-gate closure based on high-resolution structures of a single sodium channel protein. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6p6y.cif.gz | 73.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6p6y.ent.gz | 52.2 KB | Display | PDB format |
| PDBx/mmJSON format | 6p6y.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p6/6p6yftp://data.pdbj.org/pub/pdb/validation_reports/p6/6p6y | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6p6wC  6p6xC  3rvyS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29768.320 Da / Num. of mol.: 1 / Mutation: V100C, Q150C Source method: isolated from a genetically manipulated source Source: (gene. exp.) Arcobacter butzleri (strain RM4018) (bacteria)Strain: RM4018 / Gene: Abu_1752 / Production host:  Trichoplusia ni (cabbage looper) / References: UniProt: A8EVM5 Trichoplusia ni (cabbage looper) / References: UniProt: A8EVM5 | ||||||
|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 614.877 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YM Mass: 614.877 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C32H58N2O7S / Comment: detergent*YM#3: Chemical | ChemComp-PX4 / 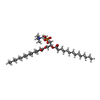  Mass: 678.940 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C36H73NO8P / Comment: DMPC, phospholipid*YM Mass: 678.940 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C36H73NO8P / Comment: DMPC, phospholipid*YMHas ligand of interest | N | Has protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 6.29 Å3/Da / Density % sol: 80.45 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 4.8 / Details: 1.8 M ammonium sulfate 0.1 M sodium citrate pH 4.8 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS / Beamline: 8.2.1 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 315r / Detector: CCD / Date: Jul 29, 2016 |
| Radiation | Monochromator: Double-crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2.89→50 Å / Num. obs: 16988 / % possible obs: 97.9 % / Redundancy: 12.9 % / Biso Wilson estimate: 54.6 Å2 / CC1/2: 1 / Rmerge(I) obs: 0.125 / Rpim(I) all: 0.035 / Rrim(I) all: 0.13 / Χ2: 1.125 / Net I/σ(I): 22.06 |
| Reflection shell | Resolution: 2.89→2.94 Å / Redundancy: 4.9 % / Rmerge(I) obs: 0.649 / Mean I/σ(I) obs: 1.21 / Num. unique obs: 638 / CC1/2: 0.831 / Rpim(I) all: 0.253 / Rrim(I) all: 0.703 / Χ2: 0.755 / % possible all: 76.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3RVY Resolution: 2.89→44.53 Å / Cor.coef. Fo:Fc: 0.925 / Cor.coef. Fo:Fc free: 0.892 / SU B: 13.046 / SU ML: 0.24 / Cross valid method: THROUGHOUT / ESU R: 0.35 / ESU R Free: 0.3 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 75.234 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.89→44.53 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|