 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6owd: Arginine Containing Reengineered Coiled-Coiled Dimer to Examine t... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6owd | ||||||
|---|---|---|---|---|---|---|---|
| Title | Arginine Containing Reengineered Coiled-Coiled Dimer to Examine the Impact of Proximal Cation Identity on Hydrophobically-Driven Assembly | ||||||
 Components Components | R+7 | ||||||
 Keywords Keywords | DE NOVO PROTEIN / Parallel coiled-coil / Dimer / De novo peptide | ||||||
| Biological species | synthetic construct (others) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.5 Å | ||||||
 Authors Authors | Biok, N.A. / Bingman, C.A. / Gellman, S.H. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: Biochemistry / Year: 2019 Title: Retention of Coiled-Coil Dimer Formation in the Absence of Ion Pairing at Positions Flanking the Hydrophobic Core. Authors: Biok, N.A. / Passow, A.D. / Wang, C. / Bingman, C.A. / Abbott, N.L. / Gellman, S.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule:  MolmilJmol/JSmol MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6owd.cif.gz | 37.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6owd.ent.gz | 26.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6owd.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ow/6owdftp://data.pdbj.org/pub/pdb/validation_reports/ow/6owd | HTTPS FTP |
|---|
-Related structure data
| Related structure data | 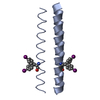 4dzmS S: Starting model for refinement |
|---|---|
| Similar structure data |





-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein/peptide | Mass: 3544.177 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) synthetic construct (others) #2: Chemical | ChemComp-CL /   Mass: 35.453 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Cl Mass: 35.453 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Cl#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 63 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.93 Å3/Da / Density % sol: 36.35 % |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 5.5 Details: No salt, 0.1 M Bis-Tris pH 5.5, 3.0 M sodium chloride |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 21-ID-D / Wavelength: 1.1272 Å |
| Detector | Type: DECTRIS EIGER X 9M / Detector: PIXEL / Date: Feb 17, 2019 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1272 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→36.59 Å / Num. obs: 8381 / % possible obs: 95 % / Redundancy: 6.7 % / Biso Wilson estimate: 32.74 Å2 / CC1/2: 0.997 / Rmerge(I) obs: 0.1088 / Rpim(I) all: 0.04586 / Rrim(I) all: 0.1184 / Net I/σ(I): 12.09 |
| Reflection shell | Resolution: 1.5→1.554 Å / Redundancy: 7 % / Rmerge(I) obs: 1.003 / Mean I/σ(I) obs: 1.34 / Num. unique obs: 808 / CC1/2: 0.424 / Rpim(I) all: 0.4111 / Rrim(I) all: 1.086 / % possible all: 93.73 |
- Processing
Processing
| Software |
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4DZM Resolution: 1.5→36.59 Å / Cross valid method: FREE R-VALUE
| ||||||||||||||||
| Displacement parameters | Biso mean: 36.62 Å2 | ||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→36.59 Å
|
