response to methotrexate / dihydrofolate reductase / dihydrofolate reductase activity / tetrahydrofolate biosynthetic process / one-carbon metabolic process / response to xenobiotic stimulus / response to antibiotic Similarity search - Function
Dihydrofolate reductase, type II / R67 dihydrofolate reductase / SH3 type barrels. - #60 / Mechanosensitive ion channel MscS, beta-domain superfamily / Electron transport accessory-like domain superfamily / SH3 type barrels. / Roll / Mainly Beta Similarity search - Domain/homology
Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.22 Å3/Da / Density % sol: 44.66 %
Crystal grow
Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: The protein was concentrated to 20 mg/mL in 100 mM Tris pH 8.0. Immediately before crystallization, chymotrypsin was added to the sample in a ratio of 1:100 chymotrypsin:protein, and the ...Details: The protein was concentrated to 20 mg/mL in 100 mM Tris pH 8.0. Immediately before crystallization, chymotrypsin was added to the sample in a ratio of 1:100 chymotrypsin:protein, and the protein was diluted to 15 mg/mL using MPD, resulting in a final MPD concentration of 25%. Reservoirs were prepared using 750 uL of 100 mM sodium phosphate pH 7.6 and 60% MPD in a Greiner 24-well hanging-drop crystallization plate. On a siliconized glass cover slip (Hampton Research), 1.5 uL of protein were combined with 2.5 uL of reservoir solution and suspended over the well. The plate was incubated at 277 K and crystals were obtained in a few days.
-
Data collection
Diffraction
Mean temperature: 93 K / Serial crystal experiment: N
Resolution: 1.4→41.09 Å / Cor.coef. Fo:Fc: 0.975 / Cor.coef. Fo:Fc free: 0.975 / SU B: 0.725 / SU ML: 0.029 / SU R Cruickshank DPI: 0.0493 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.049 / ESU R Free: 0.049 Details: Authors state that inhibitor LBA is partially modelled at the active site. Binding of the ligand has been confirmed biochemically, and ligand disorder outside of the pore center has been ...Details: Authors state that inhibitor LBA is partially modelled at the active site. Binding of the ligand has been confirmed biochemically, and ligand disorder outside of the pore center has been observed for other known ligands. More details can be found in the primary citation.
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.1577
624
5.1 %
RANDOM
Rwork
0.1412
-
-
-
obs
0.142
11543
99.62 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Canada, 5items
Canada, 5items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj




 Assembly
Assembly

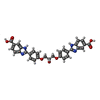
 Mass: 564.545 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H24N4O7 / Feature type: SUBJECT OF INVESTIGATION
Mass: 564.545 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C31H24N4O7 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
Mass: 118.174 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H14O2 / Comment: precipitant*YM
 Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4
Mass: 94.971 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: PO4 Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 64 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing