Component-ID: _ / Ens-ID: 1 / Beg auth comp-ID: ASP / Beg label comp-ID: ASP / End auth comp-ID: ASP / End label comp-ID: ASP / Refine code: _ / Auth seq-ID: 1 - 177 / Label seq-ID: 25 - 201
Dom-ID
Auth asym-ID
Label asym-ID
1
A
A
2
B
B
-
Components
#1: Protein
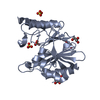
TrypsininhibitorA / Kunitz-type trypsin inhibitor A
Mass: 24031.252 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Glycine max (soybean) / Gene: KTI3 / Production host: Glycine max (soybean) / References: UniProt: P01070
Mass: 18.015 Da / Num. of mol.: 225 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.23 Å3/Da / Density % sol: 49 % / Description: thin plates
Crystal grow
Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 6.5 Details: Lyophilized protein dissolved in water to 25 mg/ml. Sitting drop vapor diffusion against reservoirs of 25% PEG 3350 with 0.10 M MES buffer ph 6.5. 3 ul drops composed of equal amounts of ...Details: Lyophilized protein dissolved in water to 25 mg/ml. Sitting drop vapor diffusion against reservoirs of 25% PEG 3350 with 0.10 M MES buffer ph 6.5. 3 ul drops composed of equal amounts of protein stock solution and reservoir supplemented with 0.10 M 1,5-Disulfonyl Naphthalene. Crystallization time about 2 weeks. PH range: 6.3 - 6.7
-
Data collection
Diffraction
Mean temperature: 173 K / Serial crystal experiment: N
Resolution: 2.4→32.61 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.909 / SU B: 32.498 / SU ML: 0.391 / Cross valid method: THROUGHOUT / ESU R: 1.697 / ESU R Free: 0.378 / Stereochemistry target values: MAXIMUM LIKELIHOOD / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.30595
656
4.9 %
RANDOM
Rwork
0.20924
-
-
-
obs
0.21376
12813
93.2 %
-
Solvent computation
Ion probe radii: 0.7 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1 Å / Solvent model: MASK
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj Assembly
Assembly


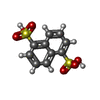
 Mass: 288.297 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H8O6S2
Mass: 288.297 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C10H8O6S2
 Mass: 195.237 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM
Mass: 195.237 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 18.015 Da / Num. of mol.: 225 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 225 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: BL7-1 / Wavelength: 1 Å
/ Beamline: BL7-1 / Wavelength: 1 Å Processing
Processing