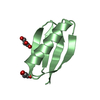
- PDB-6nlb: Crystal structure of de novo designed metal-controlled dimer of m... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6nlb
Title
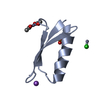
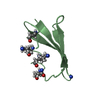
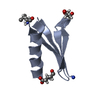
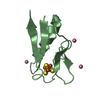
Crystal structure of de novo designed metal-controlled dimer of mutant B1 immunoglobulin-binding domain of Streptococcal Protein G (L12H, E15V, T16L, T18I, V29H, Y33H, N37L)-apo
Components
Immunoglobulin G-binding protein G
Keywords
METAL BINDING PROTEIN / Metal-mediated complex / B1 Domain of Streptococcal protein G / Immunoglobulin binding protein
#256 - Apr 2021 SARS-CoV-2 Spike and Antibodies similarity (1)
-
Assembly
Deposited unit
A: Immunoglobulin G-binding protein G B: Immunoglobulin G-binding protein G C: Immunoglobulin G-binding protein G D: Immunoglobulin G-binding protein G hetero molecules
Resolution: 2.3→37.15 Å / Cor.coef. Fo:Fc: 0.948 / Cor.coef. Fo:Fc free: 0.882 / SU B: 13.416 / SU ML: 0.2 / Cross valid method: THROUGHOUT / ESU R: 0.3 / ESU R Free: 0.066 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.25762
416
5.2 %
RANDOM
Rwork
0.18784
-
-
-
obs
0.19156
7581
85.96 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Streptococcus (bacteria)
Streptococcus (bacteria) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj

 Assembly
Assembly





 Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Mg Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 88 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing