 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6n4i: Structural basis of Nav1.7 inhibition by a gating-modifier spider... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6n4i | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural basis of Nav1.7 inhibition by a gating-modifier spider toxin | ||||||
 Components Components |
| ||||||
 Keywords Keywords | METAL TRANSPORT / sodium channel / toxin / gating-modifier / voltage-gated | ||||||
| Function / homology | sodium channel regulator activity / calcium channel regulator activity / toxin activity / lipid binding / extracellular region / Chem-6OU / Beta/omega-theraphotoxin-Tp2a Function and homology information Function and homology information | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) Thrixopelma pruriens (green velvet) Thrixopelma pruriens (green velvet) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.541 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.541 Å | ||||||
 Authors Authors | Xu, H. / Koth, C.M. / Payandeh, J. | ||||||
 Citation Citation | Journal: Cell / Year: 2019 Title: Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Authors: Hui Xu / Tianbo Li / Alexis Rohou / Christopher P Arthur / Foteini Tzakoniati / Evera Wong / Alberto Estevez / Christine Kugel / Yvonne Franke / Jun Chen / Claudio Ciferri / David H Hackos / ...Authors: Hui Xu / Tianbo Li / Alexis Rohou / Christopher P Arthur / Foteini Tzakoniati / Evera Wong / Alberto Estevez / Christine Kugel / Yvonne Franke / Jun Chen / Claudio Ciferri / David H Hackos / Christopher M Koth / Jian Payandeh /  Abstract: Voltage-gated sodium (Nav) channels are targets of disease mutations, toxins, and therapeutic drugs. Despite recent advances, the structural basis of voltage sensing, electromechanical coupling, and ...Voltage-gated sodium (Nav) channels are targets of disease mutations, toxins, and therapeutic drugs. Despite recent advances, the structural basis of voltage sensing, electromechanical coupling, and toxin modulation remains ill-defined. Protoxin-II (ProTx2) from the Peruvian green velvet tarantula is an inhibitor cystine-knot peptide and selective antagonist of the human Nav1.7 channel. Here, we visualize ProTx2 in complex with voltage-sensor domain II (VSD2) from Nav1.7 using X-ray crystallography and cryoelectron microscopy. Membrane partitioning orients ProTx2 for unfettered access to VSD2, where ProTx2 interrogates distinct features of the Nav1.7 receptor site. ProTx2 positions two basic residues into the extracellular vestibule to antagonize S4 gating-charge movement through an electrostatic mechanism. ProTx2 has trapped activated and deactivated states of VSD2, revealing a remarkable ∼10 Å translation of the S4 helix, providing a structural framework for activation gating in voltage-gated ion channels. Finally, our results deliver key templates to design selective Nav channel antagonists. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6n4i.cif.gz | 453.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6n4i.ent.gz | 374.3 KB | Display | PDB format |
| PDBx/mmJSON format | 6n4i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n4/6n4iftp://data.pdbj.org/pub/pdb/validation_reports/n4/6n4i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  0341C  0342C  6n4qC  6n4rC  5ek0S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |








-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33453.512 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host: Trichoplusia ni (cabbage looper)#2: Protein/peptide | Mass: 3839.687 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Thrixopelma pruriens (green velvet) / Production host: synthetic construct (others) / References: UniProt: P83476#3: Chemical | ChemComp-6OU / [( 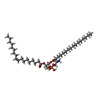  Mass: 717.996 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C39H76NO8P / Comment: phospholipid*YM Mass: 717.996 Da / Num. of mol.: 16 / Source method: obtained synthetically / Formula: C39H76NO8P / Comment: phospholipid*YMHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.71 Å3/Da / Density % sol: 73.87 % |
|---|---|
| Crystal grow | Temperature: 292 K / Method: vapor diffusion, sitting drop / pH: 7 Details: 2.3-2.6M Ammonium sulfate, 100 mM HEPES, pH 7.0; 30% sucrose for cryo PH range: 4.6-7.5 |
-Data collection
| Diffraction | Mean temperature: 80 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Nov 21, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.5→50 Å / Num. obs: 479752 / % possible obs: 99.3 % / Redundancy: 13.7 % / CC1/2: 0.999 / Rmerge(I) obs: 0.115 / Net I/σ(I): 10.43 |
| Reflection shell | Resolution: 3.5→3.59 Å / Redundancy: 13.7 % / Rmerge(I) obs: 0.76 / Num. unique obs: 35190 / CC1/2: 0.756 / % possible all: 99.6 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5EK0 Resolution: 3.541→36.892 Å / SU ML: 0.4 / Cross valid method: FREE R-VALUE / σ(F): 1.37 / Phase error: 35.23
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.541→36.892 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Origin x: 99.0797 Å / Origin y: 248.6897 Å / Origin z: 216.8423 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Selection details: all |
