Entry Database : PDB / ID : 6menTitle Crystal structure of a Tylonycteris bat coronavirus HKU4 macrodomain in complex with adenosine diphosphate glucose (ADP-glucose) Replicase polyprotein 1ab Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Method / / / Resolution : 1.5 Å Authors Hammond, R.G. / Schormann, N. / McPherson, R.L. / Leung, A.K.L. / Deivanayagam, C.C.S. / Johnson, M.A. Funding support Organization Grant number Country National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI) R35GM119456
Journal : Proc.Natl.Acad.Sci.USA / Year : 2021Title : ADP-Ribose and Analogues bound to the DeMARylating Macrodomain from the Bat Coronavirus HKU4Authors : Hammond, R.G. / Schormann, N. / McPherson, R.L. / Leung, A.K.L. / Deivanayagam, C. / Johnson, M.A. History Deposition Sep 6, 2018 Deposition site / Processing site Revision 1.0 Sep 11, 2019 Provider / Type Revision 1.1 Dec 18, 2019 Group / Category / Item Revision 1.2 Jan 13, 2021 Group / Category Item _citation.country / _citation.journal_abbrev ... _citation.country / _citation.journal_abbrev / _citation.journal_id_ASTM / _citation.journal_id_CSD / _citation.journal_id_ISSN / _citation.pdbx_database_id_DOI / _citation.title / _citation.year Revision 1.3 Oct 11, 2023 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Bat coronavirus HKU4
Bat coronavirus HKU4 X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors United States, 1items
United States, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads














 PDBj
PDBj




 Assembly
Assembly



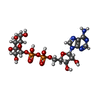
 Mass: 589.342 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H25N5O15P2
Mass: 589.342 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H25N5O15P2 Mass: 18.015 Da / Num. of mol.: 237 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 237 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing