 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6i2b: Crystal Structure of the Protein-Kinase A catalytic subunit from ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6i2b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Crystal Structure of the Protein-Kinase A catalytic subunit from Cricetulus Griseus in complex with compounds RKp153 and RKp117 | |||||||||
 Components Components |
| |||||||||
 Keywords Keywords | TRANSFERASE / Complex / peptidic ligand | |||||||||
| Function / homology |  Function and homology information Function and homology informationcAMP-dependent protein kinase inhibitor activity / histone H1-4S35 kinase activity / cAMP-dependent protein kinase / regulation of protein processing / dorsal/ventral neural tube patterning / cAMP-dependent protein kinase activity / cellular response to parathyroid hormone stimulus / protein localization to lipid droplet / regulation of bicellular tight junction assembly / cAMP-dependent protein kinase complex ...cAMP-dependent protein kinase inhibitor activity / histone H1-4S35 kinase activity / cAMP-dependent protein kinase / regulation of protein processing / dorsal/ventral neural tube patterning / cAMP-dependent protein kinase activity / cellular response to parathyroid hormone stimulus / protein localization to lipid droplet / regulation of bicellular tight junction assembly / cAMP-dependent protein kinase complex / interleukin-2-mediated signaling pathway / sperm capacitation / regulation of osteoblast differentiation / cellular response to cold / protein kinase A regulatory subunit binding / negative regulation of glycolytic process through fructose-6-phosphate / ciliary base / intracellular potassium ion homeostasis / TORC1 signaling / mesoderm formation / plasma membrane raft / axoneme / sperm flagellum / postsynaptic modulation of chemical synaptic transmission / negative regulation of TORC1 signaling / protein serine/threonine/tyrosine kinase activity / protein export from nucleus / cellular response to glucagon stimulus / cellular response to nutrient levels / positive regulation of gluconeogenesis / positive regulation of phagocytosis / acrosomal vesicle / regulation of proteasomal protein catabolic process / positive regulation of protein export from nucleus / neural tube closure / cellular response to glucose stimulus / negative regulation of smoothened signaling pathway / neuromuscular junction / positive regulation of cholesterol biosynthetic process / mRNA processing / positive regulation of insulin secretion / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / manganese ion binding / cellular response to heat / adenylate cyclase-activating G protein-coupled receptor signaling pathway / postsynapse / regulation of cell cycle / nuclear speck / protein domain specific binding / protein serine kinase activity / protein serine/threonine kinase activity / centrosome / ubiquitin protein ligase binding / protein kinase binding / perinuclear region of cytoplasm / glutamatergic synapse / magnesium ion binding / mitochondrion / ATP binding / nucleus / cytosol / cytoplasm Similarity search - Function | |||||||||
| Biological species |   Cricetulus griseus (Chinese hamster) Cricetulus griseus (Chinese hamster) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.97 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.97 Å | |||||||||
 Authors Authors | Mueller, J.M. / Heine, A. / Klebe, G. | |||||||||
| Funding support |  Germany, 1items Germany, 1items
| |||||||||
 Citation Citation | Journal: Acs Omega / Year: 2019 Title: Conceptional Design of Self-Assembling Bisubstrate-like Inhibitors of Protein Kinase A Resulting in a Boronic Acid Glutamate Linkage Authors: Mueller, J.M. / Kirschner, R. / Geyer, A. / Klebe, G. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6i2b.cif.gz | 208.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6i2b.ent.gz | 167.9 KB | Display | PDB format |
| PDBx/mmJSON format | 6i2b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i2/6i2bftp://data.pdbj.org/pub/pdb/validation_reports/i2/6i2b | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5nw8C  5o0eC  5o5mC  5ol3C  5ot3C  5ouaC  5ousC  6egwC  6eh0C  6eh2C  6em2C  6em6C  6em7C  6emaC  6ertC  6eruC  6ervC  6erwC  6esaC  6i2aC  6i2cC  6i2dC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41193.746 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Cricetulus griseus (Chinese hamster) / Tissue: Ovary / Gene: PRKACA / Organ: OvaryProduction host:  References: UniProt: P25321, cAMP-dependent protein kinase | ||||||
|---|---|---|---|---|---|---|---|
| #2: Protein/peptide | Mass: 1912.092 Da / Num. of mol.: 1 / Source method: obtained synthetically Details: PKI 5-22 with T16RBT mutation. A ribose has been covalently attached to a threonine of the peptide Source: (synth.) Cricetulus griseus (Chinese hamster) / References: UniProt: G3HK48 | ||||||
| #3: Chemical | 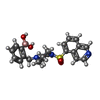  Mass: 425.309 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H24BN3O4S / Feature type: SUBJECT OF INVESTIGATION Mass: 425.309 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H24BN3O4S / Feature type: SUBJECT OF INVESTIGATION#4: Sugar | ChemComp-RIP / | 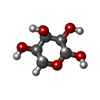  Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1 Type: D-saccharide, beta linking / Mass: 150.130 Da / Num. of mol.: 1Source method: isolated from a genetically manipulated source Formula: C5H10O5 / Feature type: SUBJECT OF INVESTIGATION #5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 95 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 95 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.63 Å3/Da / Density % sol: 53.31 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, sitting drop / pH: 6.9 Details: 100 mM Mes-Bis-Tris, 75 mM lithium chloride, 1 mM DTT, 0.1 mM sodium EDTA, 0.25 mM Mega 8, 0.7 mM peptidic ligand, 5 mM RKp117, 15% v/v methanol/water in reservoir |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Dec 10, 2016 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 1.97→48.913 Å / Num. obs: 29620 / % possible obs: 99.4 % / Redundancy: 4.83 % / CC1/2: 0.999 / Rsym value: 0.04 / Net I/σ(I): 22.78 |
| Reflection shell | Resolution: 1.97→2.09 Å / Redundancy: 4.92 % / Mean I/σ(I) obs: 3.09 / Num. unique obs: 4728 / CC1/2: 0.842 / Rsym value: 0.487 / % possible all: 98.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.97→48.913 Å / SU ML: 0.25 / Cross valid method: FREE R-VALUE / σ(F): 1.39 / Phase error: 25.65
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.97→48.913 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
