 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6i1g: Crystal structure of TP domain from Chlamydia trachomatis Penicil... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6i1g | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of TP domain from Chlamydia trachomatis Penicillin-Binding Protein 3 in complex with piperacillin | ||||||
 Components Components | Penicillin-binding protein,Penicillin-binding protein | ||||||
 Keywords Keywords | PEPTIDE BINDING PROTEIN / penicillin-binding protein / peptidoglycan / Transpeptidase | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptidoglycan glycosyltransferase / peptidoglycan glycosyltransferase activity / penicillin binding / membrane => GO:0016020 Similarity search - Function | ||||||
| Biological species |   Chlamydia trachomatis (bacteria) Chlamydia trachomatis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.13 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.13 Å | ||||||
 Authors Authors | Bellini, D. / Koekemoer, L. / Newman, H. / Dowson, C.G. | ||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: Crystal structure of TP domain from Chlamydia trachomatis Penicillin-Binding Protein 3 in complex with piperacillin Authors: Bellini, D. / Koekemoer, L. / Newman, H. / Dowson, C.G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6i1g.cif.gz | 135.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6i1g.ent.gz | 104.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6i1g.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i1/6i1gftp://data.pdbj.org/pub/pdb/validation_reports/i1/6i1g | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6hzhS S: Starting model for refinement |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 36467.785 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Chlamydia trachomatis (bacteria) / Gene: pbp_2, ERS133248_00492 / Production host: References: UniProt: A0A0E9FXJ8, peptidoglycan glycosyltransferase #2: Chemical | 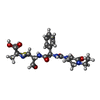  Mass: 519.571 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H29N5O7S / Feature type: SUBJECT OF INVESTIGATION Mass: 519.571 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C23H29N5O7S / Feature type: SUBJECT OF INVESTIGATION#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 17 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 17 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 53.95 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, sitting drop Details: 1 M sodium citrate and 0.1 M sodium cacodylate pH 6.5 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: Diamond / Beamline: I03 / Wavelength: 0.98 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Jul 13, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 2.13→93.36 Å / Num. obs: 21761 / % possible obs: 91.8 % / Redundancy: 11.7 % / Rrim(I) all: 0.422 / Net I/σ(I): 9.1 |
| Reflection shell | Resolution: 2.13→2.42 Å |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 6HZH Resolution: 2.13→93.36 Å / Cor.coef. Fo:Fc: 0.878 / Cor.coef. Fo:Fc free: 0.839 / SU B: 6.465 / SU ML: 0.169 / Cross valid method: THROUGHOUT / ESU R: 2.001 / ESU R Free: 0.385 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.032 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: 1 / Resolution: 2.13→93.36 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|