- PDB-6hmz: Crystal Structure of a Single-Domain Cyclophilin from Brassica na... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6hmz
Title
Crystal Structure of a Single-Domain Cyclophilin from Brassica napus Phloem Sap
Components
Cyclosporin
Peptidyl-prolyl cis-trans isomerase
Keywords
ISOMERASE / Peptidyl-Prolyl cis/trans Isomerase / Phloem Sap / CYP-like Domain / Cyclophilin Diversity / Cyclosporin A
Function / homology
Function and homology information
cyclosporin A binding / peptidylprolyl isomerase / peptidyl-prolyl cis-trans isomerase activity / protein folding / metal ion binding / cytoplasm Similarity search - Function
Mass: 18.015 Da / Num. of mol.: 50 / Source method: isolated from a natural source / Formula: H2O
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 3.07 Å3/Da / Density % sol: 59.91 % Description: Bipyramidal crystals grew to a maximum size of approximately 0.2 mm in all three dimensions within three weeks.
Crystal grow
Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7 Details: The protein concentration was adjusted to 10 mg/ml. The protein solution was supplemented with cyclosporin A at a molar ratio of 2:1 and mixed with the reservoir solution containing 2.4 M ...Details: The protein concentration was adjusted to 10 mg/ml. The protein solution was supplemented with cyclosporin A at a molar ratio of 2:1 and mixed with the reservoir solution containing 2.4 M sodium malonate, pH 7.0.
-
Data collection
Diffraction
Mean temperature: 100 K
Diffraction source
Source: SYNCHROTRON / Site: PETRA III, EMBL c/o DESY / Beamline: P14 (MX2) / Wavelength: 1.0332 Å
Detector
Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Jun 8, 2017
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.0332 Å / Relative weight: 1
Reflection
Resolution: 1.977→70.12 Å / Num. obs: 16197 / % possible obs: 99.8 % / Redundancy: 5.6 % / Rpim(I) all: 0.046 / Rrim(I) all: 0.112 / Rsym value: 0.101 / Net I/av σ(I): 6.1 / Net I/σ(I): 9.2
Reflection shell
Diffraction-ID: 1
Resolution (Å)
Redundancy (%)
Rmerge(I) obs
Mean I/σ(I) obs
Num. unique obs
Rpim(I) all
Rrim(I) all
Rsym value
% possible all
1.98-2.08
5.9
0.466
1.6
2330
0.21
0.513
0.466
100
2.08-2.21
5.6
0.372
2
2203
0.172
0.412
0.372
100
2.21-2.36
5.5
0.297
2.4
2074
0.136
0.328
0.297
99.8
2.36-2.55
5.8
0.23
3.1
1937
0.102
0.252
0.23
99.9
2.55-2.8
5.3
0.18
4
1788
0.085
0.2
0.18
99.9
2.8-3.13
5.9
0.121
5.8
1633
0.054
0.133
0.121
100
3.13-3.61
5.3
0.076
8.6
1447
0.035
0.084
0.076
99.7
3.61-4.42
5.9
0.05
12.5
1233
0.022
0.055
0.05
99.8
4.42-6.25
5.3
0.048
12.3
978
0.023
0.053
0.048
99.3
6.25-70.12
5.2
0.034
11.3
574
0.017
0.038
0.034
98.3
-
Phasing
Phasing
Method: molecular replacement
Phasing MR
Highest resolution
Lowest resolution
Rotation
36.82 Å
2.12 Å
-
Processing
Software
Name
Version
Classification
SCALA
3.3.22
datascaling
MOLREP
11.1.00
phasing
REFMAC
5.8.0103
refinement
PDB_EXTRACT
3.24
dataextraction
iMOSFLM
7.1.1
datareduction
Refinement
Method to determine structure: MOLECULAR REPLACEMENT Starting model: 4jjm
Resolution: 1.98→70.12 Å / Cor.coef. Fo:Fc: 0.963 / Cor.coef. Fo:Fc free: 0.951 / SU B: 4.747 / SU ML: 0.121 / SU R Cruickshank DPI: 0.1419 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.142 / ESU R Free: 0.137 Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS U VALUES : REFINED INDIVIDUALLY
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2231
813
5 %
RANDOM
Rwork
0.1838
-
-
-
obs
0.1856
15368
99.92 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Tolypocladium inflatum (fungus)
Tolypocladium inflatum (fungus) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Germany, 3items
Germany, 3items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
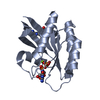

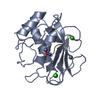
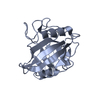



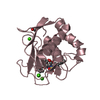
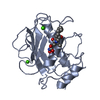

 PDBj
PDBj

 Assembly
Assembly


 Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg
Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg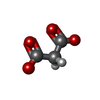
 Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4
Mass: 102.046 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H2O4 Mass: 18.015 Da / Num. of mol.: 50 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 50 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing