- PDB-6g9z: Crystal structure of human histidine triad nucleotide-binding pro... -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 6g9z
Title
Crystal structure of human histidine triad nucleotide-binding protein 1 (hHINT1) crystallized at P212121 space group, with visible extended fragment of N-terminus
Histidinetriadnucleotide-bindingprotein1 / Adenosine 5'-monophosphoramidase / Protein kinase C inhibitor 1 / Protein kinase C-interacting ...Adenosine 5'-monophosphoramidase / Protein kinase C inhibitor 1 / Protein kinase C-interacting protein 1 / PKCI-1
Mass: 13823.931 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: HINT1, HINT, PKCI1, PRKCNH1 / Plasmid: pSGA02 / Production host: Escherichia coli BL21 (bacteria) / References: UniProt: P49773, Hydrolases
Resolution: 1.43→39.52 Å / Cor.coef. Fo:Fc: 0.857 / Cor.coef. Fo:Fc free: 0.828 / SU B: 4.23 / SU ML: 0.072 / Cross valid method: THROUGHOUT / ESU R: 0.124 / ESU R Free: 0.102 / Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.27726
2041
5 %
RANDOM
Rwork
0.22362
-
-
-
obs
0.2263
38759
95.82 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj



 Assembly
Assembly

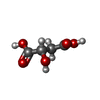
 Mass: 134.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O5
Mass: 134.087 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H6O5 Mass: 18.015 Da / Num. of mol.: 401 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 401 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 14.1 / Wavelength: 0.918409 Å
/ Beamline: 14.1 / Wavelength: 0.918409 Å Processing
Processing